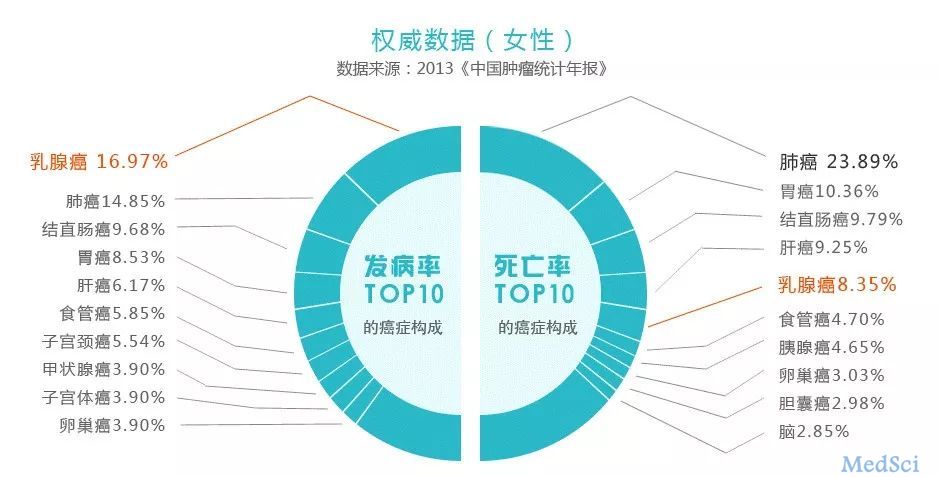
乳腺癌(breast cancer)是一类因乳腺上皮细胞增殖失控而导致的恶性肿瘤,其发病率常年位居女性恶性肿瘤首位,因此它也被称为“粉红杀手”!
在我国,乳腺癌的发病率呈逐年上升趋势,每年有30余万女性被
诊断出乳腺癌,在东部沿海地区及经济发达的大城市,乳腺癌发病率上升尤其明显。
截至目前,乳腺癌因异质性等因素,其具体病因尚未完全清楚。
所幸,随着基因组大数据分析的发展,人类对乳腺癌等恶性肿瘤相关基因突变的认知愈渐增多。
全基因组关联研究(
GWAS)已在150多个基因组区域发现与乳腺癌风险相关的基因变异。
然而,这150多个基因区域里仅有不到20个区域被详细研究,驱动这些关联的大多数变异和基因仍然是未知的。
近日,《Nature Genetics》刊发了一篇有关乳腺癌靶基因精准定位的研究论文。
该研究规模空前,由剑桥大学、哈佛大学领衔,458家
科研单位的近500位
科研人工共同完成。
研究人员利用密集基因型数据对所有已知的乳腺癌风险区域进行精细绘制,数据涉及超过217000名参与者——基因型数据来自乳腺癌协会联合会(BCAC)和BRCA1/2修饰子研究者联盟(CIMBA)。
通过人群大数据分析以及基因芯片分析,在150个乳腺癌高风险区域精准定位了191个可能的靶基因。
这些新发现的乳腺癌基因将帮助我们更详细地了解乳腺癌是如何产生和发展的,但目前已知的起作用的基因数量之多,恰恰突显了乳腺癌的复杂性!
https://doi.org/10.1038/s41588-019-0537-1
所有样本均使用OncoArray或iCOGS芯片进行基因分型,并采用逐步多元逻辑回归方法识别各区域的独立关联信号,并在每个信号中定义可信因果变量(CCVs)。
研究者发现基因组特征与CCVs有明显重叠,紧接着,他们使用贝叶斯方法,整合基因组特征和遗传关联从而精简可能的因果变量集,并计算它们的后验概率。
最后,研究人员整合了遗传、表观遗传表达以及染色质构象数据,以推断每个信号的可能靶基因。
研究流程图:
1、确定每个风险区域的可信因果变量(CCVs)
研究者对GWASs发现的150个乳腺癌风险区域进行了多次互补分析,发现了362个独立的风险信号,其中205个具有较高的可信度。
他们观察到,大多数风险区域包含多个独立信号,ESR1(雌激素受体)及其共调控基因周围区域的数量最多(9个)。
研究者使用了两种互补的方法来确定每个乳腺癌风险区域内的CCVs:
PAINTOR(一种贝斯叶方法)和传统的多项式回归方法。
CCVs支持由多项式回归发现的大多数关联,并且还确定了额外的变体。
具体来说,贝叶斯方法强调了15个很可能是因果关系的变量(HPP≥80%)。
从这些方法中,我们在每34个信号中分别识别出一个可能是因果关系的单一变量。
在其他信号中,我们也发现了四个之前被认为有害的编码变化:
无义突变rs11571833(BRCA2);
两个CHEK2编码变体(移码突变);
和剪接变异体(TERT中的rs10069690,导致端粒酶活性降低、端粒缩短和DNA损伤反应增加等)。
2、揭示CCVs在DNA水平上的功能,并预测它们的目标基因
值得注意的是,在150个乳腺癌风险区域中,许多与ER阳性(乳腺癌ER阳性率在50%~80%左右)乳腺癌风险相关的CCVs存在于标记为开放和活跃的ER阳性乳腺细胞的基因调控区域,而不存在于其他类型的细胞中。
此外,相当一部分潜在的CCVs与转录因子蛋白的结合位点以及协同调节因子相互重叠。
9种蛋白(CREBBP、EP300、ESR1、FOXI1、GATA3、MEF2B、MYC、NRIP1和TCF7L2)也出现在高可信度靶基因列表中。
这些基因编码的大多数蛋白质在雌激素信号传导通路中发挥作用。
CREBBP、EP300、ESR1、GATA3和MYC也是已知的在乳腺肿瘤中经常发生体细胞突变的癌症驱动基因。
3、与ER阳性信号相比,ER阴性信号丰富的基因组特征更少
研究者们发现相较于ER阳性信号,ER阴性信号乳腺癌关联基因更少。
BCAC和CIMBA机构的ER阴性肿瘤患者中,不到20%的基因组信号意味着更大的er阴性乳腺癌风险。
除此之外,ER阴性乳腺癌细胞系的ChIP-Seq公开数据很少和ER阴性肿瘤的异质性等因素也限制了这方面研究的展开。
尽管如此,本研究还是找寻到35个ER阴性乳腺癌可能的靶基因。
其中一些已经有功能证据支持:CASP8和MDM4。
然而,目前大多数靶点在ER阴性乳腺癌的发生发展中没有报道。
4、研究靶基因最常存在的基因本体通路
免疫、炎症和肿瘤发生之间的联系已被广泛研究,而值得注意的是,14%(25/180)的高可信度靶基因和19%的ER阴性靶基因被预测定位于
免疫系统途径中,如T细胞活化、Toll样受体级联复合物以及I-κB/ NF-κB等信号通路。
5个ER阴性高置信度靶基因(ALK, CASP8, CFLAR, ESR1 和 TNFSF10)位于I-κB/ NF-κB信号通路中,有趣的是,与ER阳性细胞相比,ER阴性细胞具有更高水平的NF-kB活性。
此外,最近对乳腺癌肿瘤组织的表达甲基化分析也发现了两个与DNA甲基化水平相关的基因簇:
一个富集于ER信号基因,另一个富集于免疫通路基因。
本研究的领导者,剑桥大学Alison Dunning博士认为,这些令人难以置信的新发现的乳腺癌基因为我们提供了更多的基因进行研究,这将帮助我们更详细地了解乳腺癌是如何产生和发展的,但目前已知的起作用的基因数量之多,恰恰突显了乳腺癌的复杂性!
总的来说,本论文的研究分析为200多个独立的乳腺癌风险信号提供了强有力的证据,确定了许多高可信度的乳腺癌靶基因。
虽然每一种基因变异只会使患乳腺癌的风险增加非常小的一部分,但综合起来,这些基因变异将让女性更清楚地了解自己的患病风险,这也将使医生和
临床医生能够就降低乳腺癌风险提供最佳策略。
参考资料:
[1] Fine-mapping of 150 breast cancer risk regions identifies 191 likely target genes
来源:BioWorld
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有,非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。本网所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。同时转载内容不代表本站立场。
在此留言