梅斯医学MedSci APP
医路相伴,成就大医



梅斯医学MedSci APP
医路相伴,成就大医
新药临床试验是为药物有效性和安全性提供客观的循证医学证据,药物的安全性, 风险收益比更是药监部门评审新药临床试验的关注点。
自2015年我国国家药品监督管理局开展新药注册申请相关的GCP 核查工作以来,安全性事件的报告,尤其是不良事件的漏报是核查中发生频次最高的不合格项目1。
为什么会存在频发的不良事件漏报,这样的漏报对药物安全性的评估会造成什么样的影响,有什么解决之道? 本界DIA年会0406分会场的专家们就业界关注的问题,分别从研究者、机构、申办方的角度对不良事件(AE)的管理做了透彻的介绍和分析,并提出改进建议;更有US FDA 检查员友情客串,从检查员角度对AE 评估和报告的重要性进行了阐述。
(1)研究者对AE 评估与报告的主体责任
药物临床试验中,研究者不但要对受试者在试验过程中发生的AE 给予足够的医疗照顾,还要根据方案、申办方和当地法规/伦理委员会的要求对发生的AE进行评估、记录、报告与跟进2。
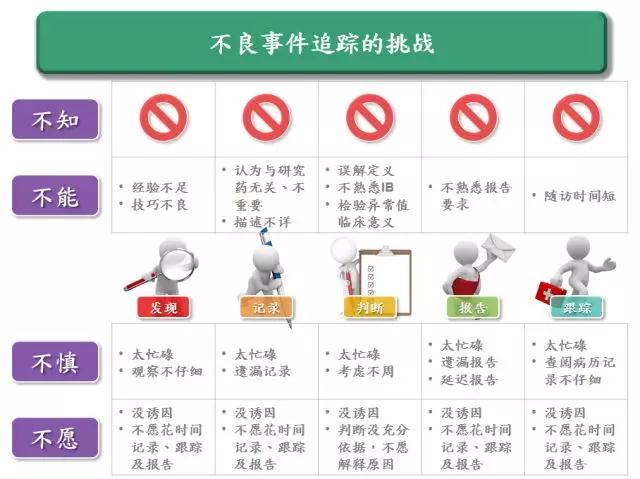
香港大学临床试验中心执行总监Henry Yau先生从临床研究中心角度分析了研究者在AE发现(identify)、记录(document)、判断(assess)、报告(report)和跟踪(follow-up)中存在缺陷的根本原因,即四个「不」(4“NOT”) ,包括不知(NOT aware)、不能(NOT capable)、不慎(NOT careful)或/和不愿(NOT willing). 从Henry的观察看,大部分研究者对GCP 关于AE 报告的要求是基本了解,即基本的awareness是有的,缺陷的根源更多的是不能、不慎和/或不愿。