梅斯医学MedSci APP
医路相伴,成就大医



梅斯医学MedSci APP
医路相伴,成就大医
表达谱芯片数据分析除了可以进行常规分析如Gene ontology 分析和Pathway分析等之外,还可以利用生物信息学软件从芯片数据中发掘更多具有深层次生物学意义的信息。目前数据深入挖掘技术主要包括:标志物预测分析(Prediction analysis for microarray)和基因网络关系分析(Gene network analysis)。这些数据分析方法同样也能应用于甲基化芯片结果和miRNA预测靶基因结果的深入挖掘。
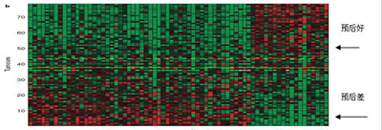

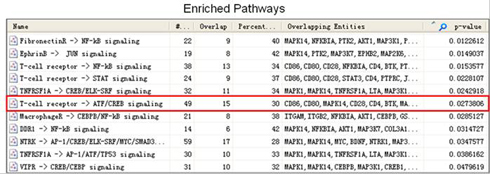
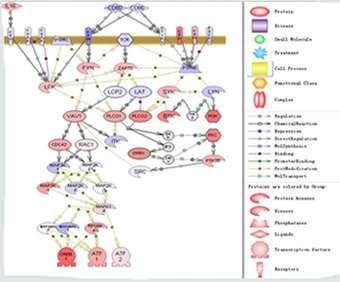
红色代表在ALL样本中上调,蓝色代表下调,颜色越深,差异表达越高;其它信息注释见图例。由此图可以看出与AML样本相比,ALL样本中T-cell receptor->ATF/CREB通路被激活。
Example2―― 关键基因分析
Pathway studio还可以从差异表达基因中找出在他们共同的调控基因。正是这些关键基因的改变,才导致众多下游基因的表达变化,维持细胞表型。找出这样的关键基因,对研究工作的进展有重要意义。
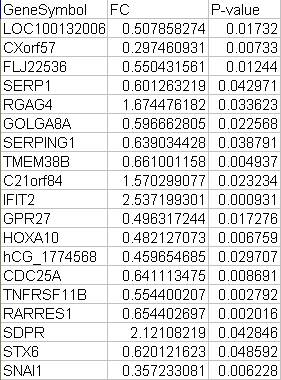
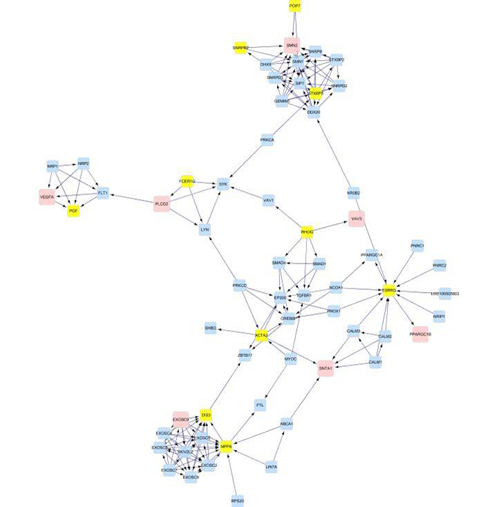
对
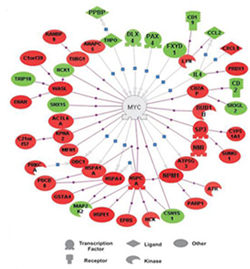
于这556个基因,利用cytoscape及其插件分析后,获得了一个由1792个节点及8320条边组成的蛋白-蛋白相互作用网络。粉红色的节点代表差
异表达基因,蓝色的节点是这些差异表达基因的第一步邻居蛋白/基因。两个节点间的边表示它们相互作用。图中孤立的节点表示没找到与其作用的蛋白。