
梅斯医学MedSci APP
医路相伴,成就大医



梅斯医学MedSci APP
医路相伴,成就大医
GCH1基因编码鸟苷-5'-三磷酸(GTP)环水解酶1(GTP-CH1),是参与四氢生物蝶呤从头生物合成的第一步和限速步骤的酶,也是黑质纹状体神经元合成多巴胺的必要辅因子。GCH1突变是导致多巴反应性肌张力障碍(DRD)的最常见原因。这种罕见的常染色体显性运动障碍主要表现为儿童时期的肢体肌张力障碍,尽管也可能存在帕金森病的特征。该病的特征是对小剂量左旋多巴没有运动并发症的发生。GCH1相关DRD患者的神经病理学和大多数多巴胺能成像研究,表明这是一种神经递质,而不是神经退行性疾病。然而,病例报告显示,一些携带GCH1突变的成人型肌张力障碍-帕金森综合征患者存在黑质纹状体多巴胺能失神经(DaTSCAN异常)。余症状可能部分源于缺乏及时治疗,暗示潜在的神经退行性变。目前还没有进行MRI研究来评估是否存在支持DRD潜在神经退行性病变的结构损伤。在这项研究中,使用磁共振成像(1.5t)来评估DRD患者与健康对照组的皮质厚度、基底节体积和白质(WM)微结构改变的模式。
9名临床表现无关的DRD患者和37名年龄匹配和性别匹配的健康对照。所有受试者均获得书面知情同意书。所有患者的DaTSCAN均正常。所有受试者均接受三维T1加权和弥散张量MRI检查。分别使用自由曲面和基于步道的空间统计分析3个大脑皮层厚度和灰质体积以及WM异常。3个具有统计学意义的结果显示p<0.05。DRD患者(年龄43.8±16.3岁;7F/2M)和健康受试者(年龄41.4±12.2岁;24F/13M)的人口统计学变量具有可比性。DRD患者(发病年龄14.2±12.1)的特点是病程长(29.2±11.9年;范围:15-46年)和长潜伏期,直到左旋多巴(159.0±225.8个月)。临床表现较轻(Burke-Fahn-Marsden评分=2.0±2.6;统一的肌张力障碍评定量表=4.9±5.7)。肌张力障碍在所有患者中都得到证实;在8名患者中作为单一症状表现,只有1例与帕金森综合征相关。皮质厚度分析显示,与对照组相比,DRD患者右侧中央前回、后扣带回、边缘上回、左侧颞中回和后扣带回皮质变薄(p=0.01-0.04)。与对照组相比,DRD患者右壳核和苍白球体积也增加(p=0.04)和苍白球体积(p=0.01)。与健康对照组相比,DRD患者皮质脊髓束、放射冠、上纵束、内囊前肢、外膜、胼胝体膝部、颞眶额WM和脑梗均有广泛的右侧WM损伤(平均和径向扩散系数增加)。
丘脑皮质和丘脑皮质下病变的皮质和丘脑皮质下的改变是相互矛盾的。这些结构影像学改变通常被解释为由于重复运动或非张力性姿势的异常感觉输入和异常运动输出而导致的可塑性改变。DRD患者显示初级运动皮质和相关感觉运动区域的皮质变薄,支持DRD中感觉运动整合过程的复杂多层次结构改变。壳核和苍白球体积增加是在局灶性肌张力障碍和无症状DYT1突变携带者中观察到的,而在帕金森病(PD)患者中发现了壳核萎缩。DRD患者病程很长,治疗开始明显延迟,但残余运动障碍非常轻微,这可能与扩大的壳核的保护作用有关,这代表着仍然保留着代偿机制。另一方面,慢性纹状体多巴胺缺乏可引起多巴胺能D2受体上调,导致基底节形态改变和肌张力障碍的发生。
形态学上的GM和WM改变表明复杂的皮质下神经网络紊乱,而不仅仅是单纯的多巴胺缺乏。然而,DRD患者的大脑形态学改变是否代表了GCH1突变的主要大脑脆弱性,还是由于疾病持续时间长和多巴胺缺乏而导致代偿机制的丧失,这个问题仍然不清楚,需要在更大的人群中进一步研究。
Dev Cell :囊胞性纤维症中粘膜下层腺体酸性pH和高浓度蛋白含量导致异常粘液
![]() 0
2020-07-30
点击查看
0
2020-07-30
点击查看
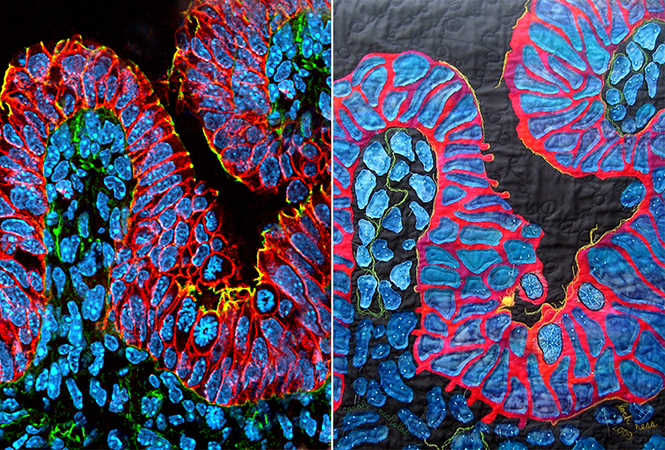
GENETICS:最恐怖食肉动物的基因突变可抑癌!或是未来人类的“救星”!
![]() 0
2020-08-11
点击查看
0
2020-08-11
点击查看
Science子刊:喝酒脸红的人,即使少量喝酒也容易患胃癌,他们都有同一个基因突变
![]() 0
2020-08-13
点击查看
0
2020-08-13
点击查看

Nature:Dantu血型如何对抗疟疾?——研究发现高张力血细胞可以抵御寄生虫入侵
![]() 0
2020-09-21
点击查看
0
2020-09-21
点击查看

Blood:灰区淋巴瘤的突变特征!
![]() 0
2020-09-28
点击查看
0
2020-09-28
点击查看

Curr Biol:睡得少又不犯困,这个让人羡慕嫉妒的基因突变,会导致自然短睡眠
![]() 0
2020-10-22
点击查看
0
2020-10-22
点击查看

