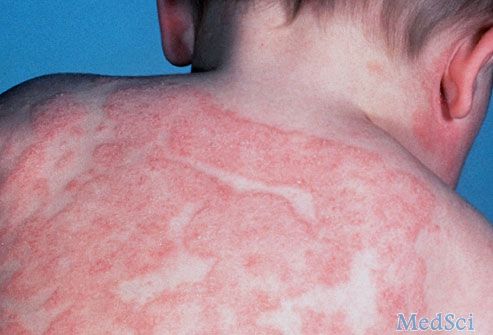
湿疹、血小板减少伴
免疫缺陷,又称Wiskott‐Aldrich syndrome(WAS),是一种原发性联合
免疫缺陷病。为X连锁隐性方式遗传,缺陷基因位于短臂11带上。本病发病机理还不清楚,血小板和血细胞表面糖蛋白(CD43)不稳定可能与免疫功能异常有关。
血小板变小,血小板减少性出血,湿疹和极易患
感染为本病的特点。体检发现肝脾肿大。血清免疫球蛋白进行性下降,以IgM尤为突出,血浆IgA、IgE水平可明显增高;血型凝集效价低下或测不出。T细胞进行性减少,T细胞功能明显减退。若不治疗,大多于3岁左右因严重出血或
感染而死亡。有人在154例WAS患儿的调查中,
诊断前仅27%患儿出现典型的三联征,即伴小血小板的血小板减少症、复发性中耳炎及湿疹。5%患儿仅有感染表现,20%仅有血小板减少症。WAS亦常伴有自身免疫性疾病。本病为少见病,预后极差,我院近20年来
诊断该病不足10例。许多医生不认识本病,易误诊或诊断为血小板减少性紫癜或湿疹。
【病例介绍】
男孩,6个月。以皮肤血肿为主诉入院。患儿于来诊前3天发热38.2℃,注射一针“退烧针”后,次日发现右臀部注射部位出现一个血肿,逐渐增大。患儿足月顺产,平素体弱,经常感冒、发热,生后反复腹泻,时有血便,各种治疗效果不佳。生后不久于头面部出现湿疹,迁延不愈,时有结痂或渗出,颈部、前胸曾有脓疱疮,当地静点“消炎药”后好转。家族史中有一位舅舅1岁左右患病夭折,据其外祖母回忆,其舅舅幼时一直患有湿疹及腹泻,时轻时重,从未治愈过,直至便血,不治身亡。
体格检查:营养发育一般,轻度贫血貌。头面、颈部、腋窝及前胸、后背均可见湿疹,毛发部可见黄色渗出性结痂,颈部可见脓疱后结痂。前胸散在出血点。心肺正常,腹软,肝肋下3cm,剑下4cm,Ⅰ°硬,脾肋下2cm,Ⅰ°硬。右臀部外上可见一个直径7cm
血肿,隆起于皮肤,无波动感,触痛明显。双足部散在瘀斑。
辅助检查:WBC 16×10 9/L,L 0﹒22,N 0﹒67,E 0﹒11,RBC 3﹒52×10 12/L,Hb 98g/L,PLT 35×10 9/L,血块退缩试验显示退缩不良。大便常规正常,隐血试验阴性,便
肠道菌群分析显示重度失调。血清IgG正常,IgA增高,IgM明显降低,IgE增高。淋巴细胞转化率明显降低(35%),E玫瑰花形成降低(46%)。
入院诊断:湿疹、血小板减少伴免疫缺陷(WAS)
【治疗措施】
1﹒入院后间断输入血小板4次,每次1单位,使血小板达到正常水平。
2﹒静脉给予头孢噻肟钠治疗感染。
3﹒口服蒙脱石散收敛、止泻,同时给予大剂量
肠道微生态制剂,调整肠道菌群。
4﹒住院期间静脉输注丙种球蛋白每周1次,连2次,每次2.5g(400mg/kg)。
经上述治疗后,患儿未再出现新鲜出血现象。住院一周后,体温转平稳,但仍有腹泻。继续治疗至腹泻好转,患儿家长要求出院,住院天数:16天。出院后未能取得联系,失去随访。
【诊治评述】
1﹒WAS是有T细胞功能缺陷和抗体应答反应低下的联合免疫缺陷病,故存在联合免疫缺陷病的共性。①生后不久即可发生感染;②对所有病原如细菌、病毒、霉菌、原虫的易感性均增高;③应注意家族中有无类似发病者。④实验室检查既有体液免疫缺陷的证据,如IgM抗体含量降低,同族血型凝激素效价低下,又有细胞免疫功能低下的表现,如淋巴细胞绝对数减少、皮肤迟发超敏反应阴性、T细胞增殖反应低下、淋巴因子测定异常等。
2﹒WAS为性连锁隐性遗传性疾病,女性携带基因,男性发病。由于该病常伴有出血现象,故在诊断时应注意与血友病相鉴别。因为本病常有免疫功能低下为其突出特征,又有血小板小和少的特点,一般不难鉴别,必要时可做凝血功能检查加以鉴别。
3﹒WAS患儿常有难治性湿疹,血小板减少及肝脾增大,有时需要与主要侵袭皮肤和内脏的组织细胞增生症急性婴儿型—勒雪综合征相鉴别。但是后者除了有特征性皮疹外,发热、贫血、肝脾大较明显,皮疹印片或骨髓穿刺病理检查可兹鉴别。
4﹒WAS的治疗包括
(1)一般治疗:加强护理,尽可能减少和防治感染,已合并感染时选用适当抗菌药物治疗。
(2)如发生严重出血或血小板数特低时,补充血小板。
(3)替代疗法:针对其免疫缺陷,补其所缺,可使免疫功能改善,但不持久。如静脉注射免疫球蛋白(IVIG),剂量为每月400mg/kg。补充细胞因子,如胸腺素、转移因子等,仅有部分疗效。
(4)免疫重建:为患儿移植免疫器官或组织,使其在患儿体内定居存活,以恢复其免疫功能。可移植含有造血
干细胞的骨髓、脐血或肝脏等等。
骨髓移植(BMT):致死性的原发免疫缺陷病是MBT的适应证。但进行BMT后可能使短期内死亡率升高。故应仔细论证其风险受益比率。从理论上讲,造血
干细胞缺陷所致的原发性免疫缺陷采用BMT是可取的。香港一个调查显示:已做骨髓移植的原发性免疫缺陷病中,大多数移植对象为严重联合免疫缺陷病(SCID)和WAS。骨髓含有丰富的造血干细胞,故骨髓移植可以重建患儿T、B细胞和单核/巨噬细胞功能。移植前需做组织配型,HLA‐D匹配尤为重要。半合子BMT需同时使用免疫抑制剂,以减少移植物受到宿主排斥或移植物抗宿主反应(Graft‐versus‐host reaction,GVHR)。采用单克隆抗体、E‐花环形成和通过分裂原吸附柱等方法排除供体骨髓中的T细胞后,再行移植可减少GVHD的发生。WAS可以选择BMT治疗,有人统计亲属HLA同源供体BMT的生存率为90%,而用单倍体同源供体或MUDs则分别为34%和65%。
(5)如无条件进行干细胞移植,可采用IVIG、
抗生素预防和脾切除术联合治疗。香港一份报告在21例WAS患儿的回顾中,14/15例接受脾切除治疗者血小板减少症得到治愈,但3例复发。部分患儿平均血小板容积(MPV)于脾切除术后暂时正常,但8~23个月后再次出现异常。脾切除后451个月内急性严重细菌感染的发病率无明显升高,可能与
抗生素及IVIG
预防有关。4例死亡,2例为大脑B细胞
淋巴瘤,1例为进行性多灶性白质脑病,1例为肺炎后严重慢性胸腔疾病。血小板计数<10×10 9/L的患儿很可能出血,而有自身免疫疾病的患儿则有发生恶性肿瘤的可能。IVIG充分支持治疗、抗生素预防及脾切除术联用,可使WAS患儿于短期集中期存活率及生活质量提高。持续感染、出血、
血管炎及过敏症状和发生
淋巴瘤等均提示应采用BMT。如免疫重建成功可明显延长患儿的生命。BMT是否降低造血系统恶性肿瘤的发生尚不清楚。
1﹒本病与其它联合免疫缺陷病一样,可以用被动免疫方法或接种灭活疫苗的方法来预防传染病,如接种灭活脊髓灰质炎疫苗等死疫苗。严禁使用减毒活疫苗,如口服脊髓灰质炎活疫苗糖丸(OPV)、卡介苗(BCG)等,以防止疫苗感染。口服OPV亦勿用于患儿父母、兄弟姐妹及家族成员,因为OPV可间接感染而致脊髓灰质炎。
2﹒与其它细胞免疫缺陷病类似,在使用
血液制品时,应注意避免或极其慎重使用含有淋巴细胞的制品,以防止供体淋巴细胞对受者发生移植物抗宿主反应,此反应发生在输注后5~20天,表现为发热、皮疹、肝脾肿大、黄疸和腹泻,甚至死于严重感染。如确需输入全血、血浆、红细胞、白细胞等制品,需经3000Rad的X射线照射处理,以抑制供体T细胞在宿主内增殖。
3﹒应做好遗传咨询,检出致病基因携带者,并给予遗传学指导。采用原位杂交、限制性片段长度多态性分析和PCR方法可以发现缺陷的基因,从而可在产前做出诊断。
4﹒在儿科门诊如遇迁延不愈的湿疹、反复腹泻患儿,要询问有无反复发生的感染,如中耳炎、皮肤感染、肺炎等,以及一些条件致病微生物感染如鹅口疮等。尤其若有注射部位出血、或
消化道出血史者,更应检测血小板,如血小板总数减少,血小板形态也较正常小者,应进一步检测体液免疫和细胞免疫,以早日确诊是否患有本病。
来源:儿科急重症与疑难病例诊治评述
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有,非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。本网所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。同时转载内容不代表本站立场。
在此留言