
梅斯医学MedSci APP
医路相伴,成就大医



梅斯医学MedSci APP
医路相伴,成就大医
法布雷病是一种罕见的X连锁遗传溶酶体贮积症,是由于GLA基因突变导致α半乳糖 苷酶A(α⁃Gal A)活性降低或完全缺乏,造成代谢底物三己糖酰基鞘脂醇(GL⁃3)及其衍生物脱乙酰基 GL-3(Lyso⁃GL⁃3)在多脏器贮积,引起多脏器病变甚至引发危及生命的并发症。由于法布雷病缺乏特异性症状,因此需结合临床表现、酶活性、生物标志物及基因检测等结果协助临床早期诊断。随着阿 加糖酶 β和阿加糖酶 α在我国获批上市,将为我国法布雷病患者带来特异性治疗的福音。本共识以 循证医学为基础,对法布雷病的临床表现、诊断方法和流程、治疗、筛查、遗传咨询与产前诊断等方面进行阐述,为推动法布雷病的规范化诊疗提供依据。
一、法布雷病概况
因位于 Xq22.1 的 GLA 基因突变,导致其编码 的α半乳糖苷酶A(α⁃galactosidase A,α⁃Gal A)活性 降低或完全缺乏,造成代谢底物三己糖酰基鞘脂醇 (globotriaosylceramides,GL⁃3)及其衍生物脱乙酰基 GL⁃3(globotriaosylsphingosine,Lyso⁃GL⁃3)在肾脏、 心脏、神经、皮肤等大量贮积,引起相应的多脏器 病变。病情严重者出现心脑血管并发症(如心力衰 竭、卒中等)或终末期肾病,甚至过早死亡。男性 患者预期寿命减少约15~20年,女性患者减少约6~ 10年 。
二、法布雷病的临床表现
法布雷病临床表现多样,常为神经、肾脏、心 脏、皮肤、胃肠道、眼等多脏器受累。 以 GL⁃3 和 Lyso⁃GL⁃3 为主的代谢产物最早在胚胎 早期贮积于组织器官。患者多在青少年时期出现 症状,并随病程进展而逐渐加重。其中,肾脏、心 脏、脑是后期主要受累脏器。
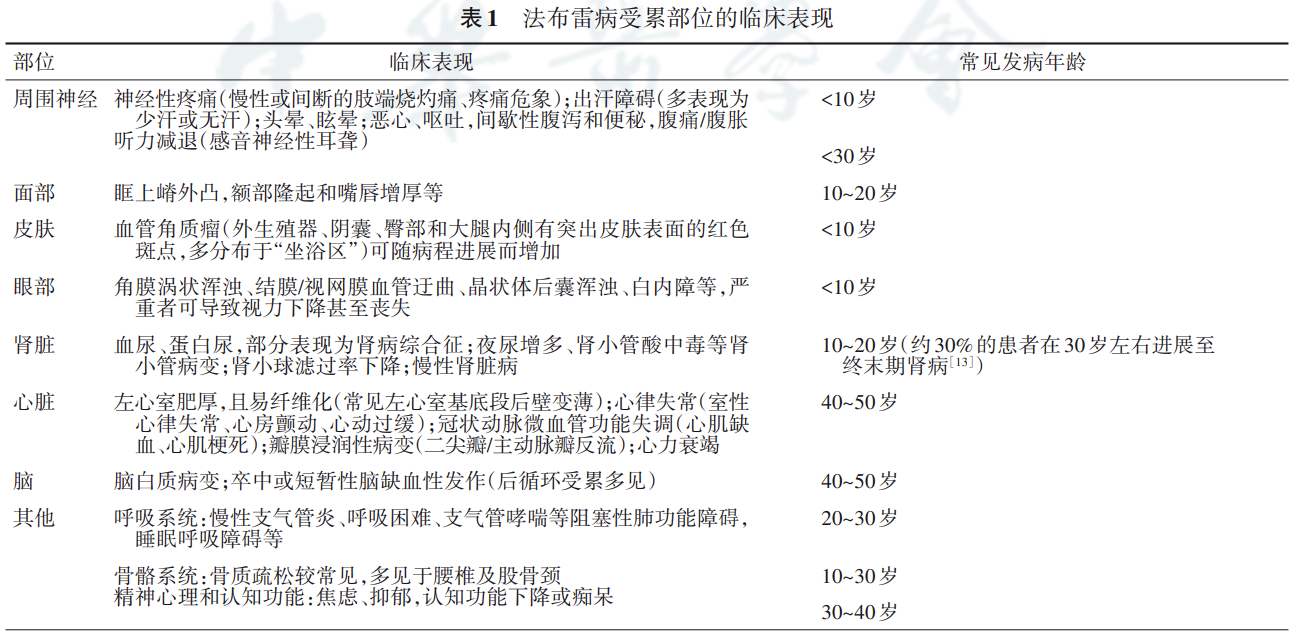

法布雷病按临床表现分为经典型和迟发型。国外文献报道,迟发型发病较经典 型≥10 倍 ,据上海交通大学医学院附属瑞金医院统计资料显示,目前国内诊断为法布雷病的患者中 66.1% 男性患者为经典型 ,75% 女性患者为迟发型。
三、法布雷病的诊断检测方法与流程
(一)诊断检测方法
1. α⁃Gal A活性检测:简单快捷,样本多为外周 血白细胞、血浆、干血纸片(DBS)等。
2. 基因检测:是诊断的重要检测手段,可提取 外周血、干血纸片样本或头发毛囊等组织的 DNA。 除明确诊断外,基因检测还能确定基因突变类型协 助判断临床表型,指导家系筛查。
3. 生物标志物检测:包括血浆GL⁃3水平和血浆 Lyso⁃GL⁃3 水平。血浆GL⁃3水平:是诊断法布雷病常用的生 化 指 标,可为 VUS 患者提供辅助诊断信息。血浆 Lyso⁃GL⁃3 水平检测敏感度比 GL⁃3 更 高 ,且与临床表型有良好的相关性。目前可通过干血纸片样本进行 Lyso⁃GL⁃3检测。
4. 组织病理学活检:具有辅助诊断意义,可 检测肾脏、心脏、皮肤或神经组织。
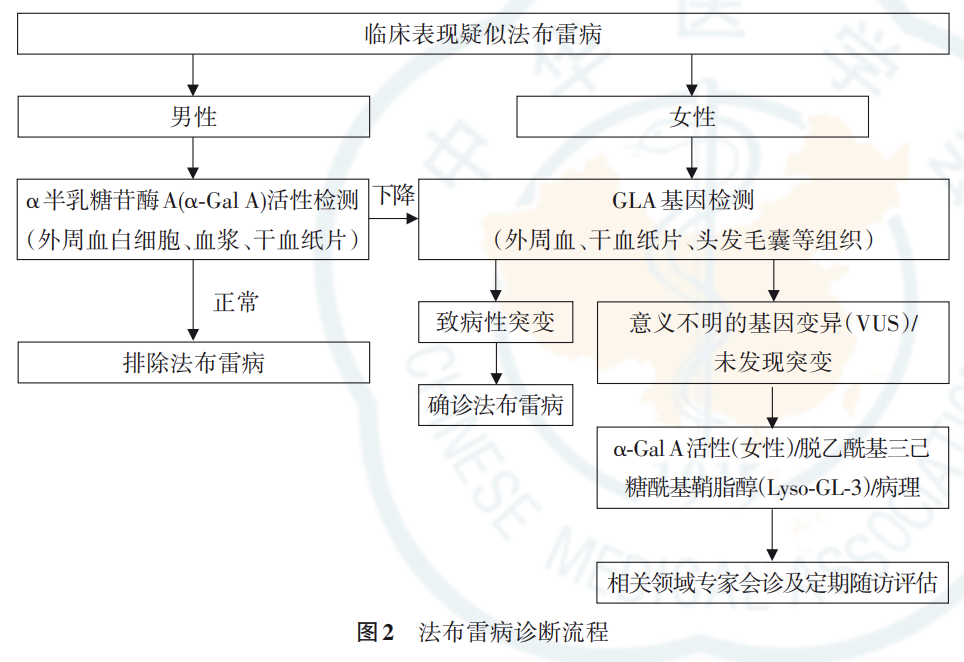
(二)诊断流程
疑似法布雷病患者的诊断流程见图 2。法布 雷病缺乏特异性症状,容易漏诊、误诊,患者出现症 状至明确诊断时间平均为 14.8 年,最长可达几十 年 。因此,法布雷病的诊断需结合临床表现、 酶活性、基因检测、生物标志物等多项指标。 诊治过程中遇到疑似患者建议转诊至有资质的医 院进行诊治。
(三)鉴别诊断
法布雷病临床表现多样且不具特异性,对无法 布雷病家族史、临床表现不典型者,诊断十分困难, 易导致误诊,需与其他疾病鉴别,如:疼痛需与风湿 免疫病、幼年特发性关节炎 、原发性红斑肢 痛症、雷诺综合征等鉴别;血管角质瘤需与过 敏性紫癜或其他皮疹鉴别;消化道症状需与消化不 良、肠易激综合征鉴别;角膜涡状浑浊需与氯喹等 药物导致的角膜浑浊等鉴别;肾脏受累需与原发 性肾小球肾炎或其他继发性肾小球疾病鉴别;心脏受累需与其他病因导致的肥厚性心肌病、 左心室肥厚 、心律失常、心力衰竭鉴别;神经系 统受累需与其他病因导致的青少年期出现的脑部 病变、早发性卒中鉴别。
四、法布雷病的治疗
法布雷病的治疗目标在于延缓疾病进展,改善 生活质量,降低相关并发症的发病率,延长患者生 存期。因此,在对患者受累脏器初步评估的基础 上,制定合适的个体化治疗方案,定期检测和调整 治疗,优化患者的疾病管理。
(一)治疗前评估
根据临床表现的严重程度差异,患者确诊后在 开始治疗前需对疾病严重度进行全面、准确的基线 评估,为确定治疗方案提供依据。

(二)治疗方法
1. ERT:ERT 通 过 外 源 性 补 充 基 因 重 组 的 α⁃Gal A,替代患者体内酶活性降低或完全缺乏的α⁃Gal A,促进 GL⁃3 的 分 解 ,减 少 GL⁃3 和 Lyso⁃GL⁃3 在器官组织的贮 积,减轻患者疼痛程度,减少蛋白 尿,并改善其他相应症状,阻止或延缓多系统病变发生。
(1)ERT的起始治疗指征:根据不同人群分为儿 童患者和成人患者,儿童患者的治疗指征, 成人患者的治疗指征。
(2)ERT 使用注意事项:ERT 相关不良反应多 见输液相关反应,大多是轻至中度,可自行缓解,或 减缓输注速度、给予非甾体抗炎药、抗组胺药和/或 糖皮质激素对输液相关反应进行处理。
此外,由于患者溶酶体缺乏α⁃Gal A活性,ERT 可导致抗药抗体的形成(主要为 IgG抗体),但关 于抗药抗体是否影响 ERT 的临床疗效尚无一致结论。
2. 对症治疗:法布雷病累及多个组织器官,对 症治疗主要是针对各脏器受累情况(具体治疗方案 需专科医生评估)。需注意的是, 仅通过对症治疗来管理法布雷病并不合适,对症治 疗并未针对潜在的法布雷病发病机制。

3. 其他治疗方法:分子伴侣疗法是一种口服小分子药物,可选择性地、可逆性地与结构、功能有缺 陷的α⁃Gal A结合,稳定蛋白构象,帮助蛋白正确折 叠以发挥正常功能,增加或恢复α⁃Gal A活性,促进 其运输至溶酶体,以清除贮积的底物,因此又称为 酶增强治疗。目前临床使用较多的是米加司 他(migalastat,已在加拿大、欧洲等国家上市,暂未 在我国上市),需注意的是,米加司他仅适用于部分 错义突变的法布雷病患者 。 此外,底物减少治疗、基因治疗及基于 mRNA 治疗等一些新的药物或治疗策略正在临床试验 或 研 发 中 ,有 望为法布雷病的治疗提供新的方向 。
(三)持续临床监测
持续监测法布雷病相关各临床参数,以保证患 者获得较好的个体化治疗,监测内容及频率。
五、筛查
1. 家系筛查:家系筛查对发现新的法布雷病患 者有重要意义。目前我国缺乏法布雷病的流行病 学资料,对确诊的法布雷病患者进行家系筛查可帮 助临床发现潜在的法布雷病患者 ,以帮助患者及 早接受 ERT 等治疗,改善预后 。国外报道,基于先证者的家系筛查可能平均检测出 3~5 名新患者 。

2. 高危人群筛查:法布雷病在肾脏疾病(慢性 肾脏病、透析)、心脏疾病(左心室肥厚、肥厚性心肌 病)、神经系统疾病(缺血性卒中)等人群中患病率更高。我国终末期肾衰竭透析患者中患病率为 0.12% 。美国血液透析患者中的患病率男性为0.21%,女性为0.15%;肾移植患者中男性为0.25%, 女性为 0;心脏病(含左心室肥厚和肥厚性心肌病) 患者中男性为0.94%,女性为0.90%;卒中患者中男 性为0.13%,女性为0.14% 。通过筛查高危人群, 可提高法布雷病的诊断率,及时采取有效治疗措施 避免严重并发症发生。干血纸片法检测高危人 群的 α⁃Gal A 活性及 Lyso⁃GL⁃3 水平,可简便、快 速、准确地早期筛查法布雷病 。
3. 新生儿筛查:新生儿筛查可通过检测 α⁃Gal A 活性、GLA 基因等,在临床症状和体征出现之前 及早发现疾病,及早开始治疗,避免出现严重并发 症或死亡发生,目前已在多个国家和地区实 施。美国伊利诺伊州和华盛顿州的新生儿中,法布 雷 病 的 发 病 率 为 1/3 000 和 1/10 000;意 大 利 为 1/3 100;奥 地 利 为 1/3 859;西 班 牙 男 性 为 1/7 575;中 国 台 湾 男 性 总 体 为 1/1 250(经 典 型 为 1/22 570,迟 发 型 为 1/1 390),女 性 为 1/40 840;日本为1/11 854。
六、遗传咨询和产前诊断
由于法布雷病是X染色体连锁遗传疾病,一般 情况下,男性患者的女性后代患病风险100%,男性 后代正常;女性患者的男性及女性后代患病风险均 为50%。因此,应对所有育龄的男性和女性患者 提供孕前和产前的遗传咨询,以及产前诊断或胚胎 植入前遗传学诊断
七、罕见病信息登记
如果您愿意寻求不断更新的信息,建议您在此登记患者的信息,即使没有完全确诊,也可以登记,点击进入:
原始出处
中国法布雷病专家协作组.中国法布雷病诊疗专家共识(2021年版).中华内科杂志 2021 年4 月第 60 卷第 4 期 Chin J Intern Med, April 2021, Vol. 60, No. 4.
PLoS One:在法布里病病人中,心脏和肾脏功能障碍与晚期听力损失相关
![]() 0
2017-11-25
点击查看
0
2017-11-25
点击查看

15年来法布里病新药Galafold获FDA加速批准
![]() 0
2018-08-11
点击查看
0
2018-08-11
点击查看

第15届年度WORLDSymposium:pegunigalsidase alfa用于治疗法布里病的初步数据
![]() 0
2019-01-29
点击查看
0
2019-01-29
点击查看

Clinica Chimica Acta:尿球蛋白-鞘氨醇及其类似物在法布里病诊断中的临床应用
![]() 0
2020-03-01
点击查看
0
2020-03-01
点击查看
Freeline的肝靶向基因疗法FLT190治疗法布里病,获得了欧洲委员会授予的孤儿药称号
![]() 0
2020-03-12
点击查看
0
2020-03-12
点击查看
Nat Commun:基因治疗:法布里病的潜在治疗策略
![]() 0
2021-03-01
点击查看
0
2021-03-01
点击查看

