
梅斯医学MedSci APP
医路相伴,成就大医



梅斯医学MedSci APP
医路相伴,成就大医
基因组结构变异(SV)通常指长度大于1Kb的基因组序列变异,主要包括缺失、插入、重复等变异类型。SV与癌症、孟德尔遗传病及其他复杂疾病相关,但对其进行检测、组装和完全解析仍具挑战性。相比短读长测序,长读长测序可检测更复杂的SV,并能有效从大的基因组中构建无参照的从头组装;但目前仍然需要采用计算方法来充分解析这些长读长测序数据,解决罕见疾病的问题。
现有调用和聚类SV的工具大多基于启发式算法,未考虑整个基因组的组装或复杂基因位点中等位基因之间的变异,从而错误地分裂或合并SV。为克服这些缺陷,必须采用基于图形的泛基因组学方法,即基因组在拓扑结构图中的相互关联。目前,由高质量基因组组装而成的图形泛基因组图谱已经被人类泛基因组参考联盟(HPRC)用于创建人类种群多样性的泛基因组参考。与线性泛基因组相比,这种图形泛基因组在检测SV方面显示出更高的灵敏度。
近日,加拿大麦吉尔大学的科研人员在Nature Communications杂志发表了题为“Pangenome graphs improve the analysis of structural variants in rare genetic diseases”的文章。研究团队探讨了在罕见病队列中使用图形泛基因组(Pangenome graphs)来表征SV的益处;利用标准参考基因组、公共组装以及罕见疾病项目(GA4K)中大量的长读长、高保真度基因组测序(HiFi-GS)数据,构建了代表GA4K统一SV调用集的图形泛基因组以识别常见变异。经验证,图形泛基因组可排除常见的非致病性或罕见的变异,并对足够罕见但具有因果关系的SV进行优先排序。

文章发表在Nature Communications
主要研究内容
一种识别和整合数百个SV的泛基因组学方法
研究团队对GA4K计划中287个父-母-子三人组(trio)进行分析,以寻找具有潜在致病性的罕见SV。在组装基因组之前,染色体微阵列分析显示该队列中超90%的先证者未被诊断,表明这组基因组富含疑难病例。为此,研究团队纳入了短读长基因组测序(srGS)亲代数据,并进一步使用PacBio HiFi Reads对所有先证者进行测序;使用HPRC发布的94个单倍体基因组扩充上述数据以鉴定、识别罕见变异;并基于minigraph构创建了一个图形泛基因组,在668个单倍体基因组和两个标准参考基因组(GRCh38和CHM13v2)中鉴定SV。
在图形构建过程中,当添加单倍体基因组时,图中缺失的大于50 bp的多态性会产生新的节点和路径。分析结果显示,每增加一个单倍体基因组,新增的非参考序列节点数量稳定在500个左右,表明在人类基因组中仍有更多的等位基因有待发现。
利用得到的图形,研究团队对组装进行了基因分型,观察到180,755个多态性位点和631,400个不同的等位基因。为确保所有基因型都来自可靠的组装序列,研究团队使用Flagger进行验证。在最佳组装中,98%的等位基因被有效区域支持。此外,研究团队还发现重复和复制序列是造成SV的主要因素。
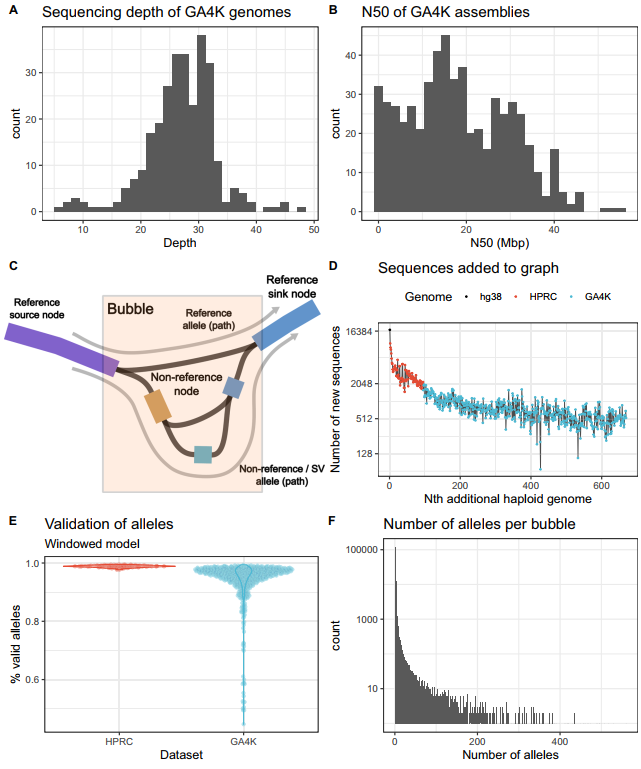
图1. 图形泛基因组的构建
基于图形泛基因组鉴定SV可以降低错误率
为证实图形泛基因组能够恢复其他长读长测序方法调用的SV等位基因,研究团队计算了在287个先证者中每个minigraph SV(基因组图谱中的非参考等位基因)的召回率和精确率,并将其与通过PBSV方法获得的SV进行比较,最终得到了一个二维分布热图。结果显示,大多数SV的精确率和召回率都较高;从整体基因型来看,minigraph的召回率为0.78,精确率为0.80。此外,与染色体微阵列分析结果相比,minigraph也达到了类似的灵敏度,在每个样本中,minigraph平均召回79.5%的染色体微阵列SV。
GA4K队列中有一对共享表型的同卵双胞胎,研究团队利用其探索了SV的复制率(即真实阳性率)。结果显示,PBSV共调用了23,516个SV,其中83.12%在两个双胞胎中被复制;minigraph调用了29,964个SV,其中84.96%被复制,表明该方法比基于参考的方法更敏感。研究团队还探索了其他58个GA4K家庭中的等位基因共享情况,发现基于minigraph的方法显示出平均比PBSV多7.1%的等位基因共享。此外,研究团队检测了GA4K232 trio中的孟德尔违反(Mendelian violations),发现minigraph相对于PBSV有更低的假阳性率和假阴性率。
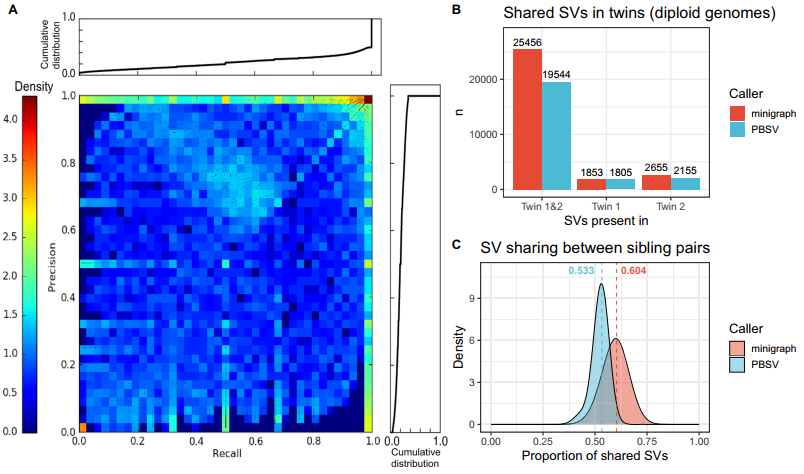
图2. 验证鉴定获得的SV
罕见SV等位基因分布在整个基因组中
研究团队利用图形泛基因组分析了可能具有功能相关性的SV等位基因,并重点研究了GA4K特有的204,551个等位基因,包括132,391个单等位基因SV,仅在GA4K中观察到一次。结果显示,在7,644个基因中共发现73,982个等位基因,其中18,095个等位基因在3,112个基因的3,772个外显子内。值得注意的是,有1,383个等位基因与275个OMIM基因的306个外显子重叠,这些基因与孟德尔遗传疾病和表型相关。
接下来,研究团队按频率划分上述等位基因,大多数是单等位基因和罕见变异,并探讨了单等位基因能否在基因组的基因间、基因中或外显子区域富集或减少。结果显示,在6,638个基因中有51,733个单等位基因SV,在2,530个基因的2,932个外显子中有13,083个,在242 OMIM基因中有978个。与预期一致,外显子和OMIM外显子的频谱对罕见等位基因的倾向性最强,其次是基因内区和基因间区。特别地,72.4%外显子SV和70.7%OMIM外显子SV为单等位基因SV。
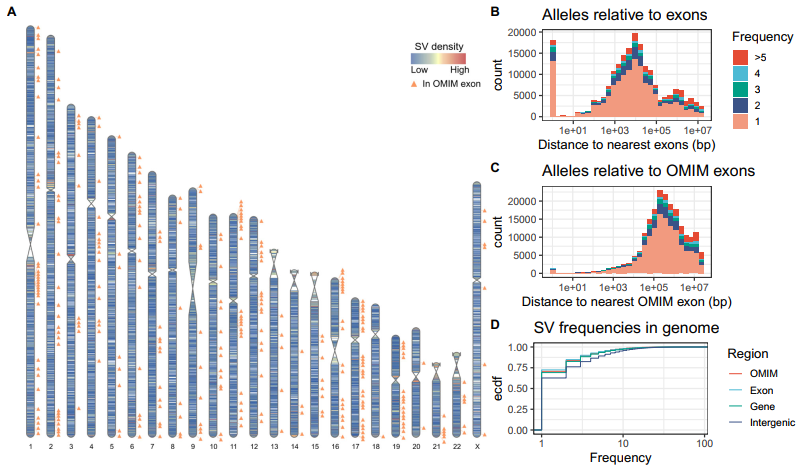
图3. 罕见SV在基因组中的分布
具有显著影响的SV
为关注具有潜在表型影响的变异,研究团队使用人类表型本体论(HPO)对每个患者的候选位点进行评分。结果显示,最终筛出40个受影响外显子的罕见SV,其中有10个出现在高多态性外显子中;在剩余30个外显子中观察到23个潜在的致病性突变,这些突变位于功能缺失(LOF)等位基因被报告为致病原因的基因中。
最后,研究团队在一名患者的母系单倍型中发现了一个新诊断性SV:在该先证者的所有疾病基因中,KMT2E基因的14.5 kbp缺失在表型匹配评分中排名前5%,并且是评分最高的外显子罕见SV,可影响KMT2E的外显子9-13。该患者具有张力减退、头部畸形和发育迟缓的神经发育表型,与KMT2E常染色体显性遗传功能变异的临床表现重叠。研究团队还通过短读长基因组数据验证了该变异,并通过PCR进行了临床验证。
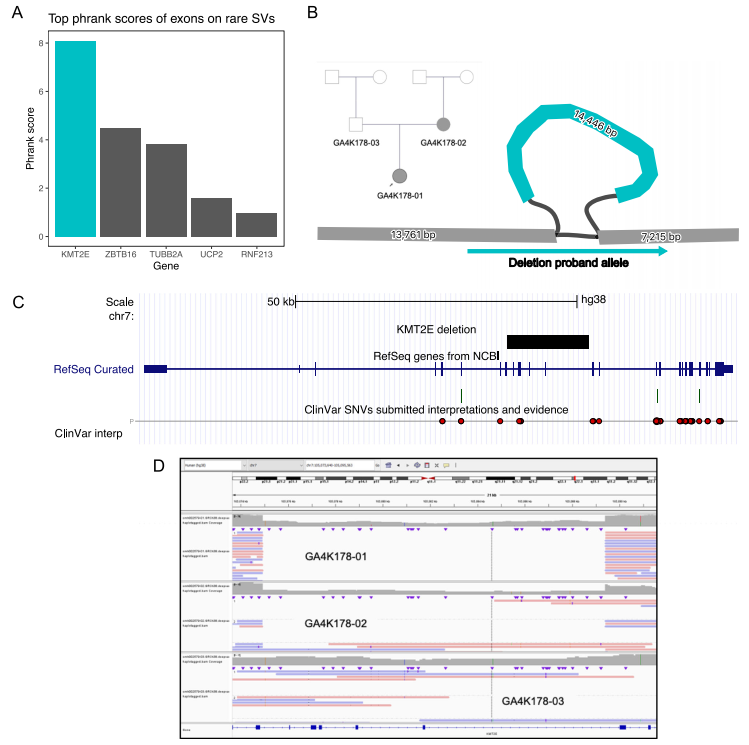
图4. KMT2E的缺失性诊断
结 语
综上所述,图形泛基因组为研究遗传变异提供了一个全面的框架,可以探索难以通过参考基因组来表征的复杂位点。研究团队基于渐进式泛基因组构建技术及大量数据构建了一个图形泛基因组。基于该方法,该研究获得了比标准参考方法更高水平的再现性,揭示了常见和罕见的SV,并对可能导致遗传病的SV进行优先排序。在未来可通过使用minigraph-cactus添加基础水平变异来扩展这种泛基因组,这将有助于揭示SV中可能存在的任何小的嵌套变异,并细化SV断点。
论文原文:
Groza, C., Schwendinger-Schreck, C., Cheung, W.A. et al. Pangenome graphs improve the analysis of structural variants in rare genetic diseases. Nat Commun 15, 657 (2024). https://doi.org/10.1038/s41467-024-44980-2.
Nat Genet:阐明基因互作模式或可帮助预测个体患病风险
![]() 0
2017-03-02
点击查看
0
2017-03-02
点击查看

亚洲家猪 “超清” 基因组图谱发布
![]() 0
2019-10-09
点击查看
0
2019-10-09
点击查看

Nat Commun:转移趋势抵抗性前列腺癌基因组图谱阐释了不同的基因型具有可能的临床影响
![]() 0
2019-12-08
点击查看
0
2019-12-08
点击查看
Nature:孙颖浩教授研究组全新发布亚洲人前列腺癌基因组图谱
![]() 0
2020-03-28
点击查看
0
2020-03-28
点击查看

Cell Res:李彦宏发表顶刊医学论文,发布很大食管鳞状细胞癌基因组图谱,为诊断和治疗奠定基础
![]() 0
2020-05-15
点击查看
0
2020-05-15
点击查看

Nat Med:仅1mL血浆即可获得癌症综合表观基因组图谱,揭示15种癌症类型的更多临床可操作信息
![]() 0
2023-11-21
点击查看
0
2023-11-21
点击查看

