梅斯医学MedSci APP
医路相伴,成就大医



梅斯医学MedSci APP
医路相伴,成就大医
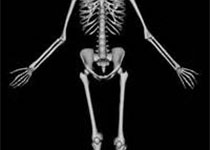
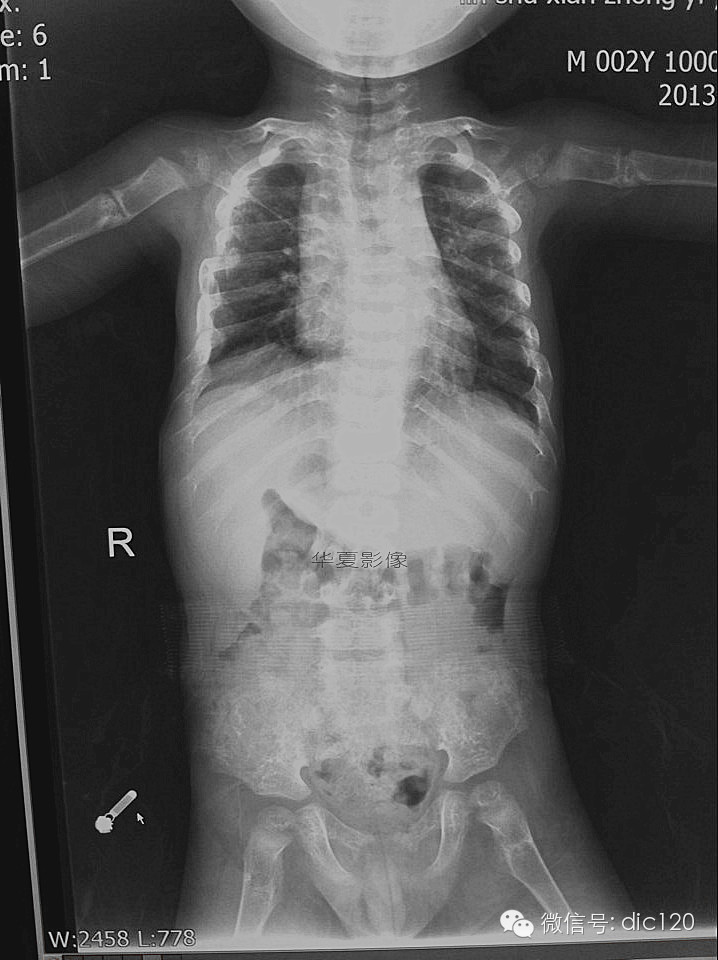
成骨不全症(Osteogenesis imperfecta,OI)是一种影响骨骼和其他器官的结缔组织疾病,具有多种遗传模式、众多致病基因和复杂的致病机制。以往的分类缺乏结构和科学依据,适用性差。
国内研究团队对成骨不全的发病机制进行了总结和梳理,并分别从I型胶原蛋白缺陷(合成缺陷、加工缺陷、翻译后修饰缺陷、折叠和交联缺陷)、骨矿化障碍、成骨细胞分化和功能缺陷等方面分析了成骨不全的分子发病机制,并总结了几种新的未分型成骨不全的致病基因及其致病机制,旨在为成骨不全症的精确诊断和治疗提供分类依据和科学依据。文章发表在《Orphanet Journal of Rare Diseases 》杂志。
过去,人们认为成骨不全仅由编码I型胶原蛋白的基因(COL1A1和COL1A2)的显性突变导致I型胶原蛋白缺陷引起。然而,随着其他罕见致病基因的发现,OI现在被认为是一种与I型胶原蛋白“相关”的疾病。罕见的致病基因参与I型胶原的翻译后修饰、加工、折叠和交联,但也参与骨矿化和成骨细胞分化。由于各亚型成骨不全的发病机制不同,其临床特征具有高度的异质性。
为了对成骨不全症进行准确的分类,团队提出了基于致病基因的成骨不全遗传分型。根据致病机制将其分为1-4型,并为其分类提供依据。
近年来,越来越多的罕见基因被发现可引起成骨不全症,这些基因的致病机制成为当前关注和研究的热点。由于不同代谢途径的改变,OI致病基因的发现和致病机制的研究可以为OI的不同形式开辟不同的药理学治疗前景。基于分子发病机制的成骨不全分类是分析患者基因型与表型相关性的基础,可以为成骨不全分类提供依据,指导临床准确诊断,预测成骨不全患者表型的严重程度,评估患者预后。
然而,仍然存在一些复杂的情况,可能无法进行准确的分类。临床医生应该认识到潜在的局限性,并结合其他临床参数来改善成骨不全的预后和监测策略。随着研究和科学技术的不断发展,成骨不全的分子分类将日益完善,其发病机制的研究将有新的突破。
参考文献:
Yu Hongjie,Li Changrong,Wu Huixiao et al. Pathogenic mechanisms of osteogenesis imperfecta, evidence for classification.[J] .Orphanet J Rare Dis, 2023, 18: 234.