
梅斯医学MedSci APP
医路相伴,成就大医



梅斯医学MedSci APP
医路相伴,成就大医
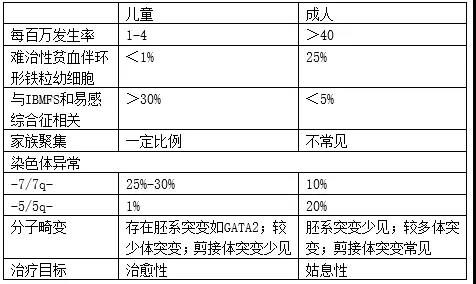

Nicotine Tob Res:二手烟入侵与喘息、鼻炎和湿疹症状之间关系研究
![]() 0
2018-03-21
点击查看
0
2018-03-21
点击查看

Ther Clin Risk Manag:城市过敏性鼻炎儿童的变化的气源性致敏原谱回顾性研究
![]() 0
2018-03-22
点击查看
0
2018-03-22
点击查看

Pediatr Allergy Immunol:中国儿童早期食物过敏和呼吸道过敏症状、注意力缺陷/多动症代表性研究
![]() 0
2018-03-23
点击查看
0
2018-03-23
点击查看

J Clin Oncol:慢性粒细胞白血病儿童新希望——达沙替尼安全有效!
![]() 0
2018-03-24
点击查看
0
2018-03-24
点击查看
Child Neuropsychol:脑疟疾对患视网膜病变的儿童在早期和中期发育影响
![]() 0
2018-03-25
点击查看
0
2018-03-25
点击查看

2018 NASPGHAN/ESPGHAN临床实践指南:儿童胃食管反流
![]() 0
2018-03-27
点击查看
0
2018-03-27
点击查看
