60岁男性,主诉是“乏力6个月,咀嚼困难4个月”。
患者6个月前肩膀和手臂肌肉感到无力,难以携带重物。4个月前开始感到吃饭时候有轻微咀嚼困难,并逐渐加重。最近3个月内自觉出现唾液分泌过多、说话不清的现象,轻微便秘,体重减轻3.6公斤。
一个月前,曾在当地县医院就诊,给予“吡啶斯的明”治疗。
外院病历显示
患者说话流畅,无构音困难,没有眼肌麻痹、上睑下垂。眼球活动自如,面部肌肉和下颌力量的测试结果正常;四肢肌力对称,肩膀外展和肘部弯曲的肌力4-5级,深部肌腱反射为3+级;血常规、电解质、B12、肝肾功能、甲状腺、肌酸激酶水平正常;梅毒抗体(-),ACh RAb和Mu SKAb双阴性;胸部CT阴性;头部MRI显示非特异轻微双侧脑白质病变;颈部MRI显示轻度颈椎病。
患者称,吡啶斯的明对他的肌无力没有作用,反而出现了新的症状。现在坐着的时候腿部无法抬起,还有频率越来越高的肌束震颤,甚至在家跌倒一次。咀嚼困难一开始有所改善,但也仍在加重。故又来大医院重新就诊。
神经科医生听完了如此复杂的自述后,以“乏力6个月,咀嚼困难4个月”将患者收治入院。
入院后先进一步采集病史,该患者的身体还真不咋样。
既往史:
高血压、银屑病、莱姆病、胸椎创伤压缩性骨折、慢性复视。
用药史:吡啶斯的明、氢氯噻嗪、厄贝沙坦、多种维生素和钙泊三醇
手术史:白内障摘除和视网膜脱离矫正
每周喝两杯酒,不抽烟。没有神经系统疾病的家族史。过敏史不详。
查体:T:36.4°C,BP:146/82 mm Hg,脉搏66次/min,呼吸16次/min,BMI:22.7。病人的精神状态正常。
颅神经检查显示:
双眼均出现向上斜视,右眼更严重;有轻微眼睑闭合无力和舌震颤。有语言性构音障碍。
四肢肌力检查可见:
颞肌、肩胛周围肌、胸肌、股四头肌、手部肌肉均可见肌萎缩,面部、胸部、大腿前侧和手臂上皆出现肌束震颤,其中手部肌束的震颤甚至使得手指自发地运动。上肢肌腱反射不对称、活跃,下肢腱反射正常对称。
感觉、共济运动和步态正常。Barbinski征(-)。
辅助检查结果:
入院后查肌酸激酶水平为498 U/L(参考范围,60~400),血清蛋白电泳结果正常。HIV阴性。
相比外院的体查结果,患者的病情加重了。
神经系统疾病鉴别
诊断的第一步就是要定位。那么,我们一步步来看。
升高的肌酸激酶水平,有可能是肌病吗?
不太可能。
肌酸激酶水平的升高并不是肌病的特异性表现,也可见于神经病变。例如,大约40%的肌萎缩侧索硬化(ALS)患者的肌酸激酶水平升高,通常是正常范围上限的1到2倍。
此外,这位患者还有肌束震颤的表现,不符合肌病的
诊断。
能定位在神经肌接头吗?
神经肌接头疾病如重症肌无力,也可累及四肢近端肌肉和延髓支配的肌肉。但是该患者几个表现不太像重症肌无力。
没有眼外肌无力,这是重症肌无力患者最常见的症状。患者没有上睑下垂,复视是慢性的,在检查中没有发现新的疲劳性眼部肌肉无力的迹象。也没有出现重症肌无力典型的晨轻暮重、病态疲劳、休息就好转的现象。
与重症肌无力相关的自身抗体检测呈阴性。虽然孤立性眼外肌麻痹的重症肌无力患者,有50%左右抗体是阴性的;但累积其他部位肌肉的重症肌无力,只有10%-20%抗体阴性。
该患者在接受吡啶斯的明治疗前就已有肌束震颤了,这是下运动神经元损伤的体征。
能定位在外周神经吗?
患者有下运动神经元性损害的体征——肌萎缩、肌束震颤,会是外周神经损害吗?
外周神经疾病所致的肌无力通常累及四肢远端肌肉,但如慢性脱髓鞘性周围神经病(CIDP)也可以累及近端。然而,外周神经病通常会有感觉障碍,与患者症状不符。
多灶性运动神经病可以累及周围神经,感觉障碍可以很轻微。然而,虽然多灶性运动神经病主要表现为进行性非对称性肢体无力,但它相对良性的进程与该患者快速进展的病情不符。
是运动神经元病吗?
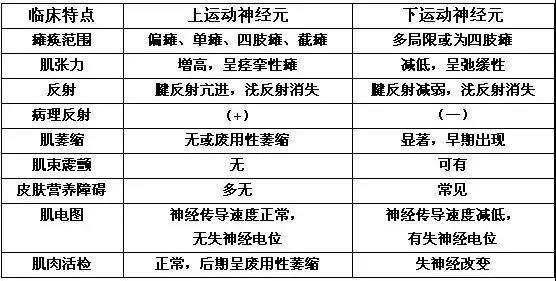
运动神经元病最常见的就是ALS。要诊断运动ALS,患者必须同时拥有上下运动神经元性瘫痪的的证据。
下运动神经元损害的证据很明显了:肌萎缩、肌束震颤。上运动神经元损害的证据呢?可以根据上肢活跃的腱反射来判断吗?
腱反射亢进的判断,通常很主观——我们不知道患者正常的情况下是什么样,没有对比,也难以判断什么样的程度算是“亢进”了。但是,对于这个病人来说,他的上肢肌肉是萎缩、震颤的,若只有下运动神经元损伤,那么腱反射应该是减弱的。在这种情况下,上肢腱反射活跃,可以作为上运动神经元损伤的证据。
除了运动神经元病,还得排除一下其他能使上肢腱反射亢进的疾病,如颈椎病、维生素B12缺乏、甲状腺功能亢进症,而从实验室检查结果来看均不考虑,那么诊断最大的指向还是——运动神经元病。另外,一种非常罕见的X染色体隐形遗传病——脊髓延髓肌萎缩症(肯尼迪病)也可以导致进行性肌无力,但其缓慢进展的病程和性征异常的体征和本例不符。
要进一步取得上运动神经元损害的证据,还有一个方法——肌电图。
于是,主治医师为病人安排了肌电图。
结果显示如下:
患者感觉神经动作电位正常,复合肌肉动作电位中上肢振幅正常,下肢振幅较低。所有传导速度均正常,排除了周围神经脱髓鞘病变。
在手臂、腿部、胸椎旁区和面部四个区域进行了肌电图。静息时,所有四个区域的肌肉都表现出束颤电位,提示下运动神经元激惹;纤颤电位和正锐波,提示失神经支配。
肌肉收缩时,记录的运动单位动作电位(MUAP)出现异常的时限延长和波幅增高,提示慢性神经再支配;MUAP呈多相波,提示新生的、不成熟的神经肌接头之间传导速度不同、放电不同步;募集相明显减少,提示脊髓前角运动神经元损害。
所有区域都出现了严重的去神经和神经再支配变化,尤其是在咬肌和上臂肌肉中。这一发现反映了患者病情的快速发展,并证实了他咀嚼困难和手臂无力的主要症状。
重复神经电刺激(3Hz)发现异常的波幅递减,脊髓副神经和面神经的幅度分别下降29.2%和11.9%。重复神经电刺激波幅衰减主要提示神经肌接头病变,但本例病人中,继发性神经肌接头病变,也即由于神经再支配所产生的不成熟神经肌接头,也可以表现出波幅递减。
总之,结合病史、
临床表现和电生理检查结果,
临床诊断为ALS。
患者接受了利鲁唑治疗——唯一
FDA批准的ALS治疗药物。在诊断后的6个月,患者病情发展迅速。他手臂完全瘫痪,腿部耐力下降,需要日常生活的帮助。出现呼吸困难和咳嗽无力,使用了辅助装置。
随着病人病情的持续发展,他开始使用无创通气装置,在诊断后1年转入社区进入临终关怀,并在临终关怀开始后3个月死亡。
在患者死亡后,他的大脑和脊髓捐献给当地医院进行研究和教学。
“世界渐冻人日”刚过去不久,ALS目前尚无很好的治愈手段,2018年5月11日,国家卫生健康委员会等联合制定的《第一批罕见病目录》将其收入其中。关注罕见病,关注渐冻人,让他们不再孤单!
来源:医学界神经病学频道
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有,非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。本网所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。同时转载内容不代表本站立场。
在此留言