
梅斯医学MedSci APP
医路相伴,成就大医



梅斯医学MedSci APP
医路相伴,成就大医
慢性中性粒细胞白血病
慢性中性粒细胞白血病(CNL)是一种少见的 BCR::ABL阴性骨髓增殖性肿瘤(MPN),其特点为成熟中性粒细胞持续增多,骨髓粒系增生并常伴有肝、脾肿大。不典型慢性粒细胞白血病(aCML)是一种费城染色体(Ph)及BCR::ABL融合基因均阴性的罕见的血液系统恶性肿瘤。
《American Journal of Hematology》近日发表一篇综述,阐述了CNL和aCML的诊断、风险分层和治疗。现翻译主要内容供参考,因为原文较长,分为CNL和aCML两个疾病分别整理,连续两天发布。

慢性中性粒细胞白血病
1.疾病概况
慢性中性粒细胞白血病(CNL)是一种罕见的BCR::ABL1阴性骨髓增殖性肿瘤(MPN),特征为成熟中性粒细胞持续增多、骨髓粒系增生和肝脾肿大,其最早记载来自1920年,随后在1964年的《柳叶刀》报告中正式定义为“CNL”。2001年,WHO的肿瘤分类采用CNL作为一个独特的临床疾病。根据2024年国家癌症研究所监测、流行病学和最终结果计划(SEER)的数据,CNL的真实发病率尚不确定,但一项基于人群的研究发现,综合SEER和国家癌症数据库数据,CNL的总发病率为0.1例/100万人,证实其罕见性。根据两个最大样本研究,诊断时的中位年龄为71岁(33-92.5岁), 56-62.5岁男性之间略多。
2.疾病特征和诊断标准
2.1临床特征
CNL是一种临床异质性疾病,多表现为偶发性中性粒细胞增多。在14例分子学定义的CNL患者中,71%的患者既往有慢性白细胞增多症,持续时间中位数为12.5个月(5-84个月)。CNL也可表现为全身症状、疲劳、骨痛、瘙痒、易瘀伤或痛风。临床检查可显示脾肿大(高达67%)和/或肝肿大,累及淋巴结少见。有数据表明CNL与出血倾向有关,包括皮肤粘膜出血、胃肠道出血,以及由血小板减少、血小板功能障碍和/或肿瘤细胞浸润引起的脑出血高发生率。早期报道中,CNL患者存在出血时间延长,二磷酸腺苷(ADP)、胶原蛋白和肾上腺素诱导的血小板聚集聚集减少,血小板腺嘌呤核苷酸含量降低。有出血倾向的患者应检测获得性血管性血友病以及其他获得性凝血和血小板功能障碍。
2.2实验室检查
CNL的特征为慢性成熟中性粒细胞慢性持续性增多。与慢性髓性白血病(CML)相比,CNL的特点为更成熟的粒细胞生成形式,分叶期(segmented stage)或杆状期(band stage)粒细胞占优势(≥80%),循环未成熟髓系前体细胞(早幼粒细胞、中幼粒细胞、晚幼粒细胞和原始细胞)很少或没有,也明显没有单核细胞增多症、嗜碱性粒细胞增多症和嗜酸性粒细胞增多症。大多数CNL患者还伴有轻度至中度贫血(中位血红蛋白[Hb] 7.9-11 g/ dL),和/或较少出现的血小板减少症。在24例WHO定义的CNL病例中,诊断时的中位外周血参数为:Hb 7.98 g/dL,白细胞计数52.7X109/L,血小板260 X109/L,中性粒细胞48 X109/L。贫血和/或血小板减少症恶化,以及白细胞增多和脾肿大,往往预示着疾病进展或原始细胞危象。乳酸脱氢酶(LDH)通常升高,白细胞碱性磷酸酶评分也升高,而CML的评分较低。由于转钴胺素从粒细胞及其前体释放,维生素B12水平经常很高。血清粒细胞集落刺激因子(G-CSF)降低在CNL患者中也有报道,但该检测并不常规进行。
2.3骨髓形态学
骨髓呈细胞过多 (>90%的细胞结构),原因在于中性粒细胞增生明显增加,骨髓与红细胞的比例增加,可能超过20:1(图1)。大多数粒细胞前体细胞处于晚幼粒细胞到分叶成熟阶段。根据定义,原始细胞<5%。通常不存在异型增生(Dysplasia)。网状蛋白染色通常正常,但也可能显示不超过1级的微小纤维化。红系成熟为正成红细胞性(normoblastic),巨核细胞虽然形态正常,但数量可能正常或略有增加。
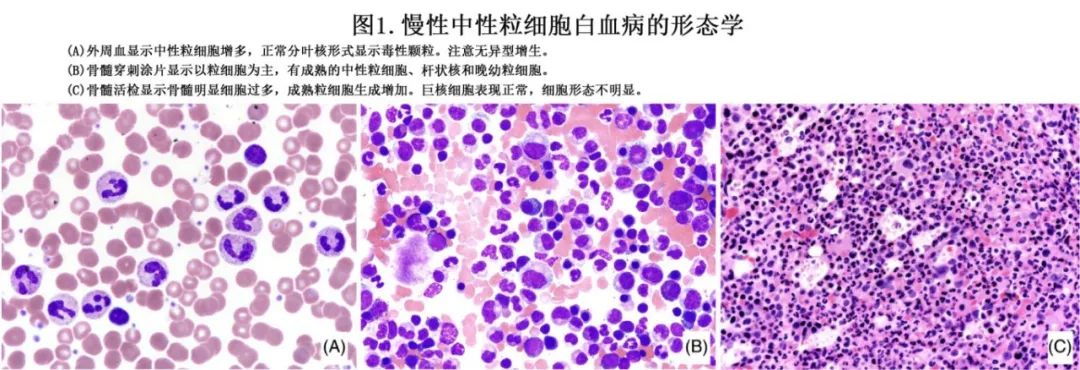
2.3.1中性粒细胞形态与功能
中性粒细胞毒性颗粒和Döhle小体虽然是非特异性的,在中性粒细胞类白血病反应中更常见,但在 CNL 中并不少见,提示中性粒细胞活化状态(图1);中性粒细胞形态也可能正常。关于中性粒细胞功能的报告与部分证明杀菌活性增加与减弱的报告不一致,尽管可能与感染倾向相关,这可能是未被重视的死亡原因。
2.4修正的诊断标准
2016年,WHO首次认可集落刺激因子3受体(CSF3R) T618I或其他激活CSF3R的突变作为CNL的关键诊断标准,且在2022年WHO第5版分类及主要诊断标准中保持不变。必须标准包括:外周血白细胞≥25X109/L;分页性中性粒细胞加上杆状形式占外周白细胞(WBC)≥80%和中性粒细胞前体细胞(早幼粒细胞、中幼粒细胞和晚幼粒细胞)占WBC的<10%,伴罕见原始粒细胞;单核细胞<10%外周白细胞,绝对单核细胞增多不符合慢性髓细胞白血病(CMML)标准,无粒细胞生成障碍。骨髓呈高细胞性,中性粒细胞比例和数量增加,呈现正常成熟,原始粒细胞<5%。排除条件包括无反应性中性粒细胞和其他MPN和MDS/MPN肿瘤;因此,不符合WHO标准的情况包括:BCR::ABL1阳性CML、真性红细胞增多症(PV)、原发性血小板增多症(ET)或原发性骨髓纤维化(PMF);PDGFRA、PDGFRB或FGFR1中不存在疾病定义性重排,并且在嗜酸性粒细胞增多和酪氨酸激酶(TK)融合的髓系/淋系肿瘤中没有PCM1::JAK2或其他融合。存在CSF3R T618I或另一种活化CSF3R的突变仍是基本诊断要求,但如果得到中性粒细胞持续增多(至少3个月)、脾肿大和无基础反应性原因(包括无浆细胞肿瘤)的支持,WHO也认可CSF3R 阴性疾病。如果存在基础浆细胞疾病,必须通过细胞遗传学/分子分析证实髓系克隆性,以进行额外的 CNL 诊断。尽管不存在 CSF3R 突变并不排除 CNL 的可能性,但应及时仔细审查诊断。
2022年 CNL 的国际共识分类 (ICC) 提案与 WHO 的总体一致。作为关键区别,ICC将白细胞增多的诊断阈值降低到≥13X109/L(如果出现 CSF3RT618I 或其他激活CSF3R的突变);但保留阈值≥25 X109/L(如果缺乏突变)。外周血和骨髓结果与 WHO 一致,ICC还明确表明,循环或骨髓原始细胞数量增加 (10–19%) 伴进行性脾肿大和血小板减少症恶化表明进入加速期;≥20%的原始细胞定义为急变期。2022年 WHO 和 ICC 的 CNL 诊断标准总结见表1。

2.5鉴别诊断
中性白细胞增多可能与多种疾病有关,包括良性和恶性。准确的 CNL 诊断需要排除潜在的混杂疾病,如反应性中性粒细胞增多/类白血病反应、CML、中性粒细胞CML(CML-N)、MDS/MPN重叠疾病如 aCML 和CMML,以及其他髓系肿瘤。
区分 CNL 与类白血病反应可能具有挑战性,因为两者均可能表现为显著的中性粒细胞增多、骨髓细胞增多,细胞遗传学正常,不存在BCR::ABL1融合基因。类白细胞反应的平均和最大 WBC 计数可能较低(某报道中分别为37.7X109/L和88.0X109/L),但计数也可能高达 100X109/L。详细的病史采集和全面的临床检查对于排除隐匿性恶性肿瘤或感染至关重要。如果发现克隆性,包括鉴定 CSF3R 突变或其他分子学或细胞遗传学异常,可支持 CNL 的诊断。
CML与 BCR::ABL1重排相关,表现为中幼粒细胞比例更高,嗜碱性粒细胞增多频繁,更常表现为血小板增多症和/或嗜酸性粒细胞增多症。虽然无费城染色体是确诊 CNL 的先决条件,但也曾报道一种罕见的CML,称为中性粒细胞CML(CML-N),其特征与 CNL 重叠,包括明显的中性粒细胞增多。CML-N的特征为不常见BCR::ABL易位,导致 e19/a2 型BCR::::ABL信使 RNA 转录,产生230 kD BCR::ABL蛋白 (p230)。临床上,与 CNL 相比,CML-N表现为总 WBC 计数降低、贫血不太严重、脾肿大不太常见/严重,原始细胞转化延迟。
不典型 CML 是一种罕见的异质性髓系肿瘤,具有 MDS 和 MPN 的共同特征。与 CNL 相反,aCML的特征为前期异型增生的中性粒细胞增多,中性粒细胞前体占 WBC 的≥10%。对比 CNL 和 aCML 的WHO 和 ICC 标准见表1。
CMML 是另一种 MDS/MPN 重叠疾病,其慢性单核细胞增多和异型增生的特征可区分其与CNL。WHO定义的 CMML 先决条件标准包括持续绝对 (≥0.5X109/L) 和相对 (≥10%) 外周单核细胞增多症,以及血液和骨髓中原始细胞<20%,不符合CML/其他 MPN 或伴 TK 融合的髓系/淋系肿瘤。支持标准包括≥1个髓系的异型增生,存在获得性克隆细胞遗传学或分子学异常(如果单核细胞增多症低于1X109/L,则需要满足两个标准),以及外周单核细胞亚群的异常分区。根据ICC,存在符合 MDS 阈值的血细胞减少症和通过异常细胞遗传学存在克隆性或存在突变的额外标准,后者需要变体等位基因频率 (VAF) 至少为10%;在没有后者的情况下,单核细胞增多症要求更严格(≥1.0 X109/L和 >10% WBC)伴原始细胞增加或形态学异型增生,或异常免疫表型符合CMML。
CNL 的诊断还需要排除其他髓系恶性肿瘤,包括PV、ET和PMF,其特征性分子学和形态学特征均在2022年 WHO 和 ICC 分类中明确定义。
罕见情况下,副肿瘤性白细胞增多可能是由于实体瘤异位产生G-CSF,类似于 CNL 的中性粒细胞增多。已报告此类病例与以下恶性肿瘤相关:包括泌尿、肺、间皮、甲状腺和肉瘤等,可能与更具侵袭性的肿瘤细胞增殖有关,可能是由于G-CSF 在自分泌生长刺激中的作用。
2.6相关疾病
2.6.1真性红细胞增多症
个别报告认为 CNL 与 PV 相关,但在几乎所有病例中,患者都暴露于 PV 的降细胞治疗,从而提出后者可能促成 CNL 发病的可能性。目前数据太有限,无法得出关于假定相关性的结论。
2.6.2浆细胞疾病
越来越多的证据表明 CNL 和浆细胞肿瘤可共存,这些病例暂时定义为“单克隆丙种球蛋白病相关CNL”(MG-CNL),高达32%的CNL患者并发浆细胞恶液质。研究发现49例“CNL”或中性粒细胞白血病样反应患者伴发多发性骨髓瘤 (MM) 或意义未明的单克隆丙种球蛋白病 (MGUS)。大多数相关病例中的浆细胞病为MM,其次为MGUS,而浆细胞瘤罕见。大多数患者表达λ轻链,剩下为κ或两者(罕见)。尚不清楚 CNL 和浆细胞疾病是否为克隆相关,或中性粒细胞增多症是否继发于基础浆细胞肿瘤。基于额外的确证性数据(包括证实肿瘤浆细胞合成 G-CSF 的研究、自发性 CNL 缓解病例和治疗异常蛋白血症后中性粒细胞计数改善),Bain和 Ahmad支持后一种假设。此外,在5例浆细胞肿瘤相关“CNL”病例中未发现单一 CSF3R 突变,这得到另一报告的支持,该报告也记录了1例浆细胞肿瘤相关 CNL 患者为CSF3R 阴性;有趣的是,发现后者与 ASXL1 和 JAK2 的畸变相关。另一份报告描述了1例IgAλ MGUS患者,表现为中性粒细胞增多、脾肿大和血清 G-CSF 升高,所有这些症状对 Janus 激酶 (JAK) 抑制剂 (JAKi) 治疗无反应,但最终对硼替佐米-地塞米松治疗有反应。有趣的是,在 CNL 检查中,发现患者在CSF3R(p.R583H和p.T640I)上携带2个罕见的单核苷酸多态性 (SNP),预测其无害,但在这种情况下意义未知。从而引起单克隆丙种球蛋白病和中性粒细胞增多症之间因果关系的可能性,高水平 G-CSF 暴露是否可促进继发性克隆性骨髓疾病的发生受到质疑。从预后角度来看,MGUS相关CNL 与孤立性CNL相比具有更长的预期寿命,因此也支持其病因不同。但其他研究对这一结论提出挑战,包括在随访期间发生 CSF3R 突变 MGUS 患者。在5例中性粒细胞增多症伴伴随浆细胞肿瘤中的2例中发现SETBP1 突变(D868N和G870S)。由于 SETBP1 突变对髓系肿瘤具有高度特异性,其发生在与单克隆丙种球蛋白病相关中性粒细胞增多症背景下,支持两个独立克隆的共存。因此,克隆性和非克隆性(反应性)中性粒细胞增多的情况可能与浆细胞肿瘤相关。总之,这些数据提醒 CNL 伴浆细胞病的真实诊断需要正式记录骨髓克隆性,并支持在这种情况下使用血清 G-CSF 水平来帮助区分反应性中性粒细胞增多。此外,浆细胞恶液质治疗在减少 G-CSF 生成和改善这些罕见情况下的临床特征(中性粒细胞增多症和/或脾肿大)方面具有潜在疗效。
2.6.3其他相关疾病
已报道 CNL 与 Sweet 综合征、自身免疫性溶血性贫血之间具有相关性,有趣的是,报告了一例继发于 CML 的CNL(CSF3RT618I和W818*突变)。后者出现治疗耐药BCR::ABL1克隆和无 ABL1 突变的 CSF3R 突变克隆扩增,推测是在具有复杂克隆结构的患者接受靶向 TKI治疗的选择压力下发生的。
2.7细胞遗传学和克隆性
在诊断和/或克隆演变过程中,少数 CNL 患者报告了细胞遗传学异常。在2002年的综述中,37%的 CNL 病例出现细胞遗传学学异常,主要包括8三体、21三体、11q缺失和 20q 缺失(最常见的异常)。随后,40例患者的 CNL 系列在32.5%(13/40) 的患者中发现细胞遗传学异常;基线时在20%的患者中发现这些异常,剩余的12.5%的患者的克隆演变过程中发现异常,包括 20q 缺失、21三体、11q缺失和 12p 缺失。一项当代研究评估了24例CNL,仅在1例患者 (4.8%) 中发现异常细胞遗传学。较少见的异常包括del(7q)、17号零体、21四倍体、7、8和9三体、1:20易位、Y缺失、6号缺失、5号增加、15号缺失和2号单体;它们在髓系恶性肿瘤中是非特异性但非随机的发现。最后,最近一份病例报告在1例 CNL 患者详细描述了一种新的KMT2A:: DCP1B 重排。由于存在于较小的亚克隆中,因此假设是与疾病进展相关的后期事件,但其致癌机制仍不清楚。
3.分子病理学(略)
4.预后和自然病程
总体预后变化较大,但多数较差。在早期的报告中,中位生存期为21至30个月,2002年的一项综述中,5年生存率为28%。2017年,一组16例CNL患者均携带CSF3R T618I突变,平均生存期为24个月,与Elliott等人报道的23.5个月相似。当代队列与历史队列相比,似乎也很少有生存期的改善,最近的两项研究显示中位生存期为15.2个月(n=24)和23.1个月(n=294)。CNL的潜在致命并发症包括颅内出血、感染、疾病进展、转化为AML或ALL,以及化疗诱导或移植后的治疗相关毒性。
ICC认为不同的CNL疾病阶段类似于CML的加速期和急变期,因为其自然历史通常概括未经治疗的CML。疾病的进展通常预示着对治疗的耐药、进行性难治性中性粒细胞增多、贫血和/或血小板减少症的恶化、器官肿大的增加、获得额外的分子学或细胞遗传学异常,以及最终的原细胞危象。白血病转化率在10%至25%之间变化,转化为AML的中位时间为15-21个月(3-94个月)。进展为PV或CMML(19例CSF3R突变患者中发生率为11%)也可能发生,但不太常见。
4.1预后变量和风险分层
历史上使用表型特征如白细胞和血小板计数来广泛预测CNL,但越来越多的可用基因数据及其相关的预后价值大大改革了风险分类。在16例 WHO 定义的 CNL 患者的研究中, WBC> 50X109/L对预后有害(中位 OS 为11个月vs. 39个月)。在14例 CSF3R 突变CNL的研究中,多因素分析显示 ASXL1 突变和血小板减少具有显著的生存劣势。尽管有报道认为SETBP1 突变参与疾病转化和治疗耐药,但在该研究中它们对生存没有影响,当代一项meta分析也未发现影响。
利用来自梅奥诊所的19例分子注释CSF3R突变CNL患者的数据,建立了预测长期生存的可操作风险模型。多因素分析建立了预后相关变量,并归因于相对风险:血小板<160X109/L(2分),白细胞>60X109/L (1分),存在ASXL1突变(1分)。低危(0-1分;N=9)和 (2-4分;n=
10)的中位OS分别为未达到和22.4个月(p=0.0016)。基于这些数据,建议高危人群进行更密切的监测,并尽早考虑进行造血干细胞移植(HSCT)。该研究还证实了CSF3R T618I与其他CSF3R变体相比对生存的明显负面影响,从分子角度支持 T618I 突变体可能代表最终 CNL 疾病的概念。
最近的基因组分析确定了 CNL 中其他预后相关的组合突变模式,NRAS、ASXL1、GATA2和 DNMT3A 突变聚集的OS有缩短的趋势;而CBL突变似乎预示着更有利的生存期。一项评估 CNL 和 aCML 队列中突变特征的预后意义的大型国际多中心研究,最近在两个疾病中发现一组6个“高危”变体:CBL、CEBPA、EZH2、NRAS、TET2和U2AF1;有趣的是,既往研究中发现的 ASXL1 的不良生存效应未得到证实。除了现有的风险范例外,在评价 CNL 的整体预后前景时,也可以考虑这些最近披露的高危突变特征。
5.治疗
目前还没有治疗CNL的标准策略,除了HSCT(仅限于少数符合条件的患者)外,没有任何治疗显示出有意义的疾病改善作用以转化为生存获益,仍存在主要的未满足需求。尽管由于缺乏循证指南和有限的治疗手段,管理仍然具有挑战性,但关于预后生物标志物、克隆进化和新的药物靶点的新数据,以及包括CNL队列在内的更强大的临床试验,是改善这种罕见疾病结局的关键步骤。
5.1常规治疗
自20世纪70年代以来,脾照射和脾切除已被用于缓解CNL的症状性脾肿大,但脾切除后粒细胞增多恶化,不应在临床实践中使用脾切除。使用口服化疗来控制CNL的白细胞减少可以追溯到20世纪70年代,尽管缓解较为短暂。羟基脲是CNL中最常用的药物,高达75%的患者表现出初始缓解(白细胞减少和/或脾肿大),中位治疗持续时间为12个月(6-87)。在梅奥诊所2018年19例CNL患者的研究中,大多数患者接受羟基脲作为一线药物治疗(82%),并且在整个病程中最终所有患者(100%)都接受羟基脲治疗;但大多数患者需要二线治疗(53%),近三分之一(32%)的患者需要三线或更多线治疗。在这个系列中使用的其他药物包括干扰素- α (IFN-a)、去甲基化药物(HMA)、芦可替尼、沙利度胺、克拉屈滨和伊马替尼。最近的一项研究类似地记录了81.5%的患者接受羟脲治疗后接受芦可替尼,重申其应优先使用作为一线药物。
IFN-a在CNL中也有很长的使用历史,并且是唯一一种提供持久缓解潜力的药物,在有限的病例报告中发表。Meyer等人描述了两例进展性CNL患者接受IFN-a治疗,分别在治疗16个月和26个月后获得长期缓解。一例患者在停用IFN后病情进展缓慢,但从未需要再次治疗(随访90个月),而另一例患者病情保持稳定(随访66个月)。其他研究类似地报告了在CNL中使用IFN-a10或聚乙二醇干扰素的长期血液学和临床缓解,持续时间为24至41个月以上。因此,干扰素仍是一种安全有效的CNL药物,应考虑作为育龄患者的一线治疗,或作为既往治疗失败的二线或后续治疗药物。
5.2JAK抑制剂
芦可替尼是一种JAK1/2抑制剂,目前批准用于治疗中高危骨髓纤维化患者、对羟基脲不耐受或难治性PV患者和难治性急性移植物抗宿主病。由于芦可替尼可抑制致癌性JAK-STAT通路信号,认为可为CNL提供临床获益。虽然未获FDA批准用于CNL,但芦可替尼已在许多小鼠模型和CSF3R t618i突变CNL和aCML患者中进行评估。Maxson等人的原始研究纳入一例携带CSF3R T618I突变的患者,该患者使用芦可替尼治疗获得临床缓解,并在治疗11个月后维持。Fleishman等人在表达CSF3R t618i的小鼠模型中证实芦可替尼的有益作用,显示细胞增殖减少,白细胞计数、脾脏重量和体重改善。在19例CNL患者中,4例(均曾暴露于羟基脲)接受芦可替尼作为二线治疗(n=3)或三线治疗(n=1)。1例的治疗正在进行中,缓解良好,但治疗时间短(2个月);另外2例中最初缓解,但最终白细胞增多症恶化并需要额外治疗(缓解持续时间分别为9.5个月和36个月);1例在急变期接受芦可替尼仅0.5个月作为移植的桥接治疗,最终获得良好的结局,并在诊断后46个月的末次随访中存活。
Dao等于2020年发表了一项备受期待的II期前瞻性多中心临床试验,该试验评估了芦可替尼对CNL (n=21)和aCML (n=23)患者的安全性和有效性。总体缓解率(ORR)为35%,包括CNL队列中的4例完全缓解和9例部分缓解,85%的患者符合至少一种临床获益的标准。CNL (与aCML相比)和CSF3R突变的存在与芦可替尼的临床反应(通过控制白细胞增多和脾脏体积减少来定义)显著相关。值得注意的是,应答者比无应答者的中位生存时间更长(23.1个月对15.6个月)。
一项针对MDS/MPN和CNL患者(NCT05177211)的JAK2抑制剂fedratinib(用于高危MF的JAK2抑制剂)的2期研究报告了初步数据。入组患者包括4例CNL, 1例aCML, 4例MDS/MPN-RS-T (MDS/MPN伴环状铁母细胞和血小板增多),1例MDS/MPN-U。患者多突变,3+个突变占70%。在5例可评估的患者中,3例(60%)在第24周缓解:3例症状缓解(75%),1例脾脏缓解(20%)(1例两者都有)。在出现显著基线症状的患者中(n=5),接受12周治疗后4例(80%)出现改善(平均-43%)。Fedratinib的安全性与既往经验一致。与芦可替尼相比,fedratinib具有更广泛的激酶抑制谱,此外还抑制FLT3和BRD4并抑制c-Myc表达,所有病例中fedratinib治疗均可降低c-Myc表达产物(平均51%,p=0.02)。
JAK抑制剂治疗对CSF3R等位基因负荷的影响已在多个研究探索,但结果相互矛盾。有的报告没有效果,有的报告混合或不一致的效果,或等位基因负荷净减少,但与症状改善没有相关性。
5.2.1影响JAK抑制剂治疗反应的变量
芦可替尼临床反应可变,推测至少部分是由于并发突变。研究评估了芦可替尼在共表达CSF3R和SETBP1的CNL中的作用(预后不良的组合),但结果不一致:Lasho等和Ammatuna分别显示芦可替尼在CNL和aCML中难治,而Nooruddin等和Stahl等报告的缓解持续时间为5至9个月。重要的是,在小鼠CNL模型中,化合物CSF3R突变已证明对芦可替尼具有耐药性,而Hinze等人的一份病例报告显示,在接受芦可替尼治疗后,携带化合物CSF3R突变(T618I和Q749X)的71岁男性患者的血液学缓解持续时间>3年,该患者此前接受过达沙替尼治疗但无效。
Stoner等在一项研究中探讨了CNL在接受JAKi治疗时特异性克隆演变的机制。显著的发现包括STAT3突变在芦可替尼治疗过程后期出现,推测对JAKi耐药有贡献,以及在疾病进展中检测到RUNX1和STAG2突变(各n=3)。后一项发现认为RUNX1和CSF3R突变在CNL进展/白血病转化中具有协同作用,并揭示了STAG2突变作为疾病进展的晚期生物标志物的潜在价值。然而,也许该研究揭示的最实际的治疗启示在于,可能需要靶向其他分子 (例如STAT3、 RUNX1),以克服耐药相关通路并根除CNL中的恶性克隆/亚克隆。
虽然还需要更多的研究来评估JAK抑制剂治疗对CNL自然史的影响,但治疗方案的缺乏和令人鼓舞的初步临床数据支持将芦可替尼或fedratinib作为不适合HSCT的CNL患者的治疗方案。未来需要解决的具体问题包括:JAKi的选择和起始时间,多线治疗的排序(一线与后续用药),以及JAKi在HSCT前的作用。由于大量数据已经在一线治疗方案失败的情况下评估了JAKi治疗,因此作为二线或后续药物,它是最具循证证据的策略。虽然没有研究专门评估JAKi在CNL围移植期的应用,但早期数据和从其他MPN(如骨髓纤维化)的经验推断,可以证明它们在这种情况下的使用是合理的,特别是在需要立即治疗白细胞增多症和/或控制症状的患者中。
5.3其他靶向治疗
CSF3R截断突变主要通过SRC激酶起作用,在体外对达沙替尼表现出药物敏感性,因此理论上支持CNL中使用SRC激酶抑制剂达沙替尼,但很少有数据表明达沙替尼在CNL患者体内具有活性。值得注意的是,最近的一项研究描述了一例携带CSF3R截断突变的B-ALL患者对化疗加达沙替尼具有良好反应。但据推测,在CNL中经常发生额外膜近端突变的背景下,达沙替尼可能不足以诱导缓解。证实这一点的是一例复合CSF3R突变的CNL患者(T618I和Q749X突变)对达沙替尼难治,最终需要芦可替尼挽救治疗。因此,达沙替尼在体内的疗效尚未得到证实,特别是在同时存在截断和膜近端突变的情况下,需要进一步研究。虽然在单一截断突变的CNL患者中进行达沙替尼试验可能是合理的,但应确保密切监测缺乏或丧失反应。
同样,在CNL患者亚群中发现NRAS突变,导致人们认为使用曲美替尼(trametinib)等药物抑制MEK可能会带来临床获益。临床前数据证明,曲美替尼在体外可抑制表达近端或复合CSF3R突变的小鼠白血病发展,虽然它在aCML的特定病例中有使用,但尚未在临床实践中进行评估。
5.4诱导化疗
标准诱导“7 + 3”化疗未显示诱导CNL血液学缓解。有报道称,在AML诱导化疗后,一例加速期CNL患者进入第二慢性期,但大多数患者要么难治,要么发生治疗相关死亡。
5.5新药治疗
最近在CSF3R t618i转化的Ba/F3细胞系中研究了aurora(极光)激酶抑制的细胞和分子学效应,对极光激酶A和B的两种抑制剂以及逆转录酶(激酶A和B的双重抑制剂)进行评估。AZD1152-HQPA(选择性极光激酶B)和reversine(双重抑制)显示出抗肿瘤潜力,导致细胞活力、克隆原性和增殖能力下降,表明极光激酶抑制在CNL中具有新的治疗潜力。目前没有其他研究药物正在进行CNL的研究。
5.6造血干细胞移植
异基因造血干细胞移植是CNL的唯一治愈手段,但大多数患者不适合移植,关于CNL中HSCT的文献仍然很少。1996年,Hasle等发表了第一份报告,记录了两例长期缓解,此后仅有有限案例证实其价值。CNL疾病的所有阶段均有移植。正如预期的那样,在加速期移植的患者结局较差,化疗方案相关的毒性增加和/或早期复发。在对CNL移植结果的回顾性分析中,,71%的患者在慢性期接受HSCT,较加速期接受移植的患者获得更持久的缓解。一项当代日本的alloo-HSCT回顾性研究收集了2003年至2014年间移植的5例CNL和14例aCML患者的数据。大多数患者接受清骨髓预处理方案。移植主要来自替代供者(n=14)和HLA相合亲缘(n=5)。CNL和aCML的一年OS率分别为40%和54.4%,考虑到标准治疗通常预后不佳,支持异基因造血干细胞移植作为其具有生存优势的实践。这项大规模的回顾性研究显示,接受一线异基因造血干细胞移植的CNL患者的长期生存率较高,两例移植患者在5年后仍存活(均<65岁;无主要合并症)。
总的来说,考虑到目前可用的治疗方法的有限疗效和通常快速致命的病程,建议考虑适合移植的CNL患者进行HSCT,特别是如果表现出高危特征。在HSCT后,监测CSF3R突变作为疾病复发的生物标志物可能是合理的。既往研究支持了这一点,研究表明,在CNL中成功进行异基因造血干细胞移植后无法检测到突变,而在aCML移植后复发的病例中,CSF3R T618I标记物持续存在。虽然需要更多的研究来确定HSCT的最佳时机,但有令人信服的证据表明,早期干预(在慢性期)有利于改善移植后结局。造血干细胞移植克服不良分子学特征的能力,药物在控制围移植期疾病中的作用,以及非清髓性预处理和替代干细胞来源的使用,都是需要进一步考虑的问题。
5.7整体管理
一种CNL管理算法如图2所示。一旦确诊,建议进行基于NGS的筛查,以识别潜在的预后有害突变,通常定义为ASXL1和SETBP1,也包括最近报道的CBL、CEBPA、EZH2、NRAS、TET2和U2AF1。
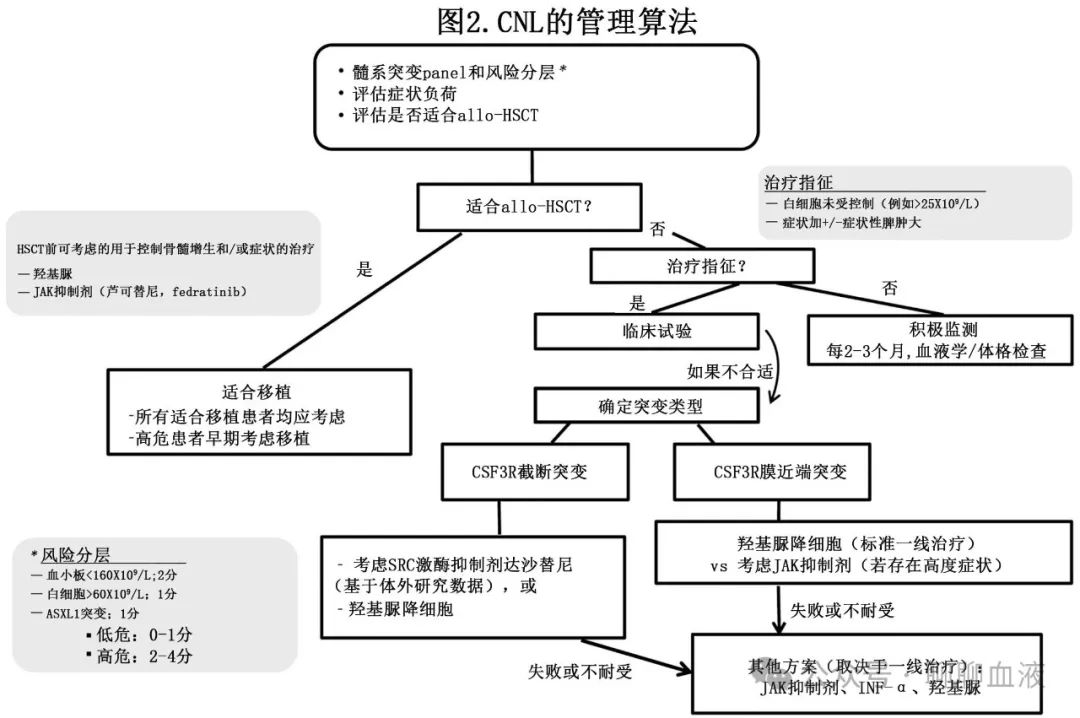
根据前文的梅奥诊所模型进行风险分层,同时对高危突变进行整体评估,可能有助于区分低危和高危患者。适合移植患者应进行异基因造血干细胞移植评估。所有患者,无论是否有移植意向和/或未来的移植计划,都应接受仔细的临床评估和监测(2-3个月或更频繁的血液学/体格检查,取决于疾病的发展速度)。对于不适合移植的患者,应优先进行临床试验。在任何时间点,骨髓增生失控(推荐的目标白细胞计数<25-30X109/L)和/或相关症状均应开始药物治疗。
参考文献
Szuber N, Orazi A, Tefferi A. Chronic neutrophilic leukemia and atypical chronic myeloid leukemia: 2024 update on diagnosis, genetics, risk stratification, and management. Am J Hematol. 2024;1‐28. doi:10.1002/ajh.27321
