梅斯医学MedSci APP
医路相伴,成就大医



梅斯医学MedSci APP
医路相伴,成就大医
病史介绍
男性,61岁,因“检查发现双侧肾上腺肿物5+天”入院,伴头昏,双侧腰部轻微不适。
辅助检查
上腹部CT显示双侧肾上腺见团片状软组织占位性病变,较大者位于左侧,大小约71x37mm,增强扫描呈轻度-中度强化(图1)。脾脏体积增大。
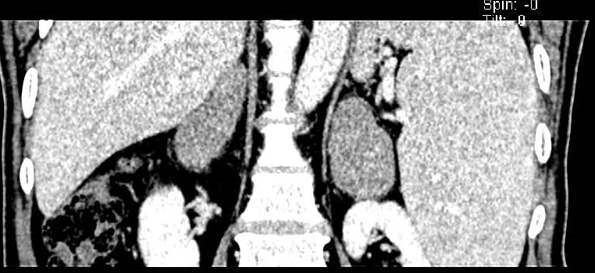
图1 上腹部CT显示双侧肾上腺见团片状软组织占位性病变,增强扫描呈轻度-中度强化。
组织病理活检
肉眼观:
灰白穿刺组织三条,长13-15mm。
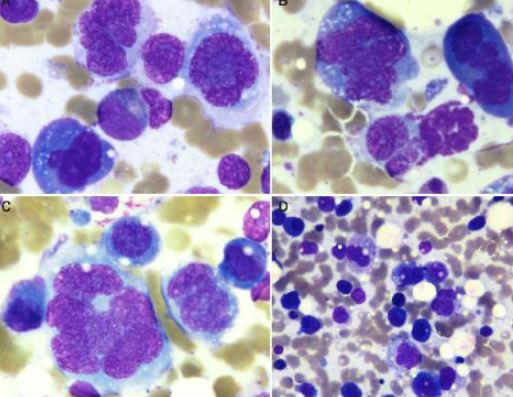
镜下观:(图2-图5)
肾上腺组织萎缩,未见明显的肾上腺皮质和髓质结构,穿刺组织中见巢团状样的上皮样细胞结构,细胞体积较大,圆形、多边形,胞浆中等,淡染,核大,空泡状,可见核仁及核分裂像,上皮样细胞团周围间质疏松水肿,纤维增生伴炎症细胞细胞浸润。肿瘤细胞之间粘附性较差,大或中等淋巴瘤样细胞,空泡状,可见核仁,呈中心母细胞样、免疫母细胞样;肿瘤细胞似乎都在一个不规则腔隙中,腔隙上皮细胞类似血管内皮细胞。
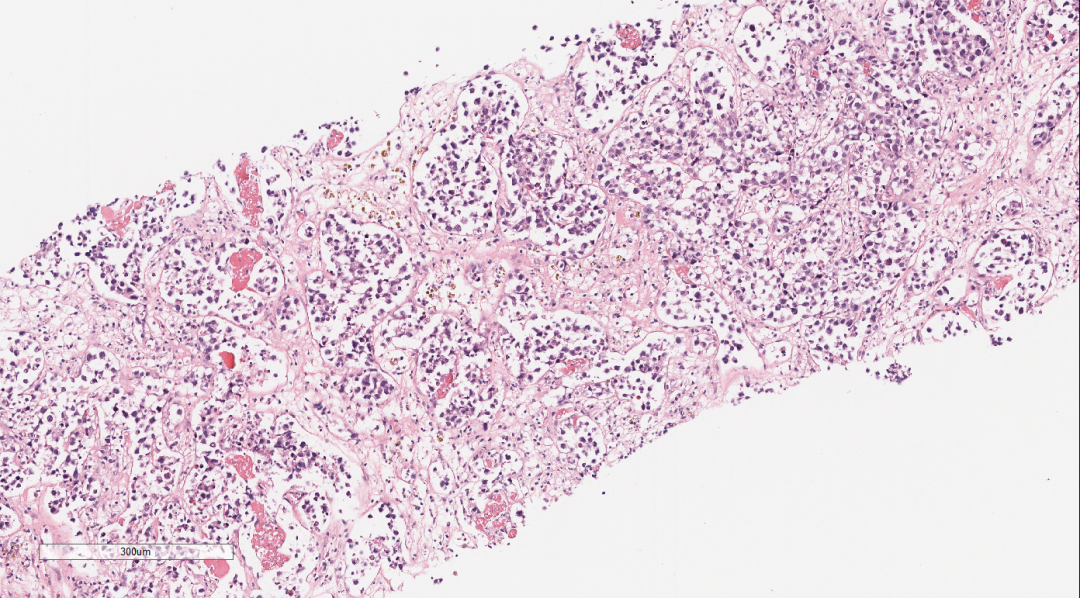
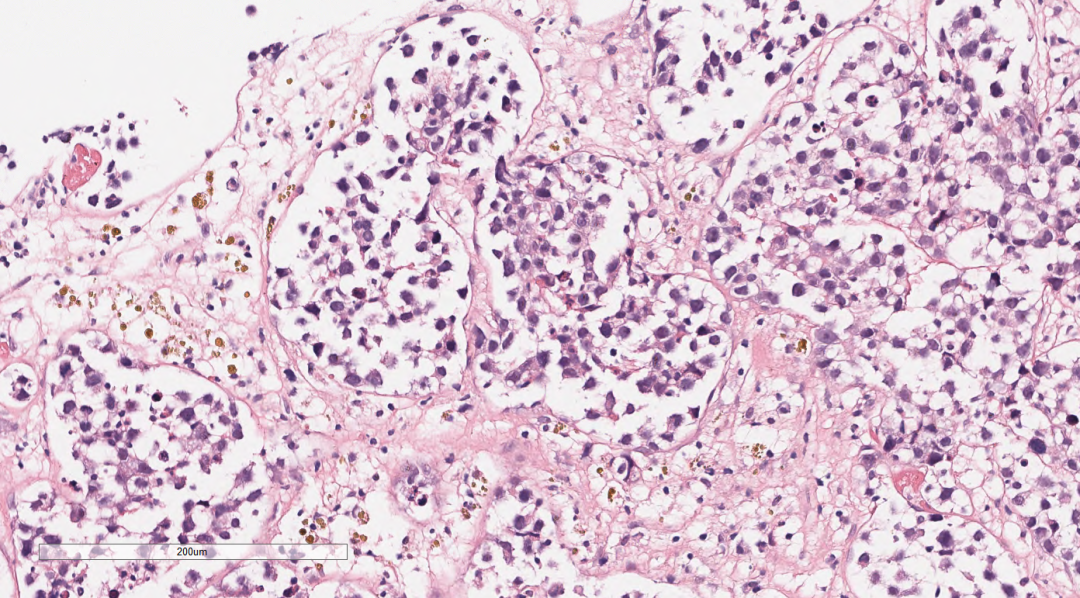
图2-图5 肾上腺组织萎缩,穿刺组织中见巢团状样的上皮样细胞结构,细胞体积较大,圆形、多边形,胞浆中等,淡染,核大,空泡状,可见核仁及核分裂像,上皮样细胞团周围间质疏松水肿,纤维增生伴炎症细胞细胞浸润。肿瘤细胞之间粘附性较差,大或中等淋巴瘤样细胞,空泡状,可见核仁,呈中心母细胞样、免疫母细胞样;肿瘤细胞似乎都在一个不规则腔隙中,腔隙上皮细胞类似血管内皮细胞。
免疫组化
肿瘤细胞Vimentin阳性,LCA(图6)、CD20(图7)、CD19、PAX-5弥漫强阳性,Ki67指数90%(图8),BcL-6(+)、MUM-1(+)、CD3(-)、CD5(-)、CyclinD1(-)、SOX-11(-)、CD10(-)、ALK(-)、CD30(-)、CD15(-)、D2-40(-),CK、EMA、a-Inhibin、Calretinin、CgA、Syn、NSE、S100、CEA、CD15、PLAP、OCT3/4、SALL4、CD117均阴性,腔隙样结构血管内皮标志物CD31、CD34阳性,肿瘤细胞完全位于CD34勾画的的血管腔隙内(图9),HHV-8阴性,EBER分子检测为阴性。
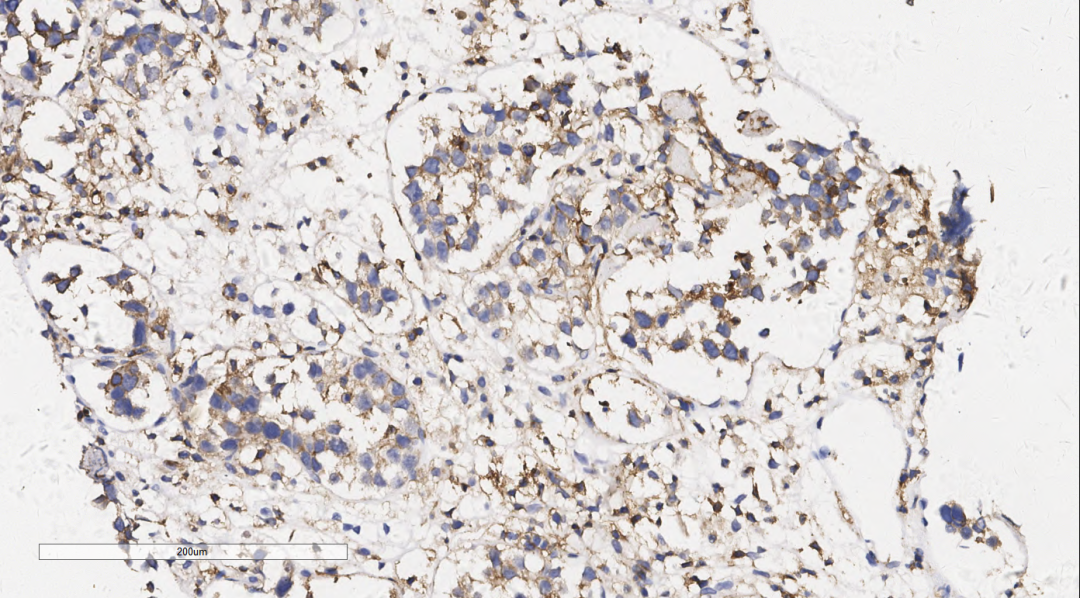
图6 免疫组化示肿瘤细胞LCA阳性。
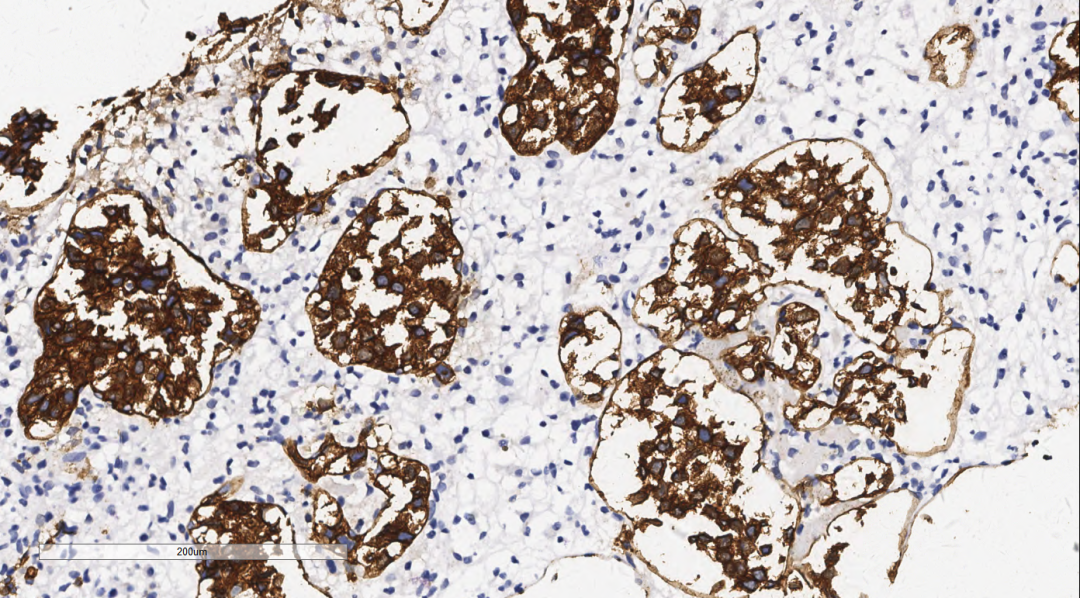
图7 免疫组化示肿瘤细胞CD20阳性。
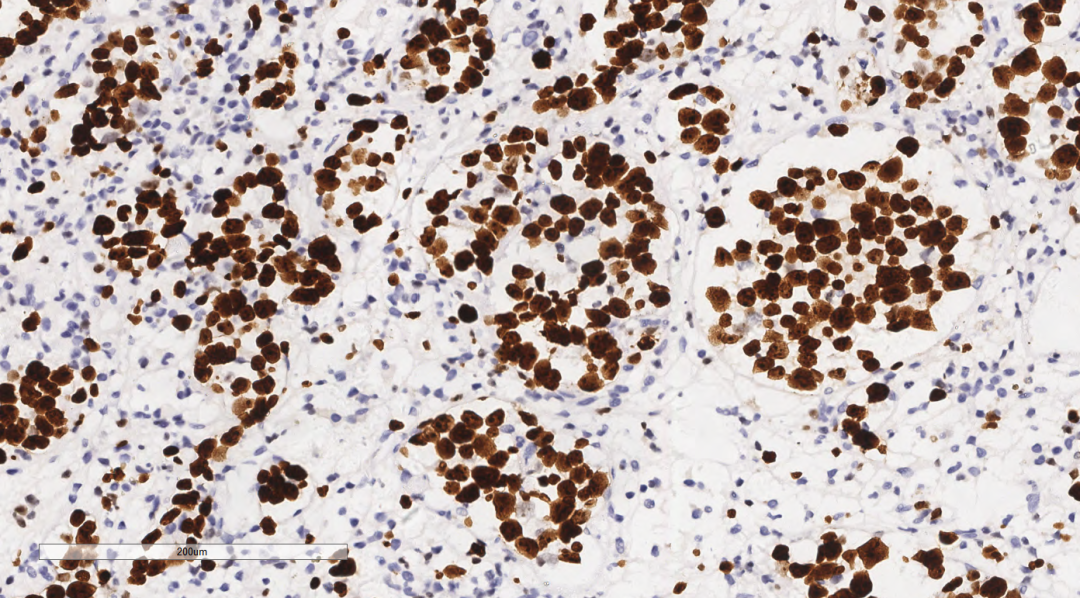
图8 免疫组化示结节内Ki67增值指数90%。
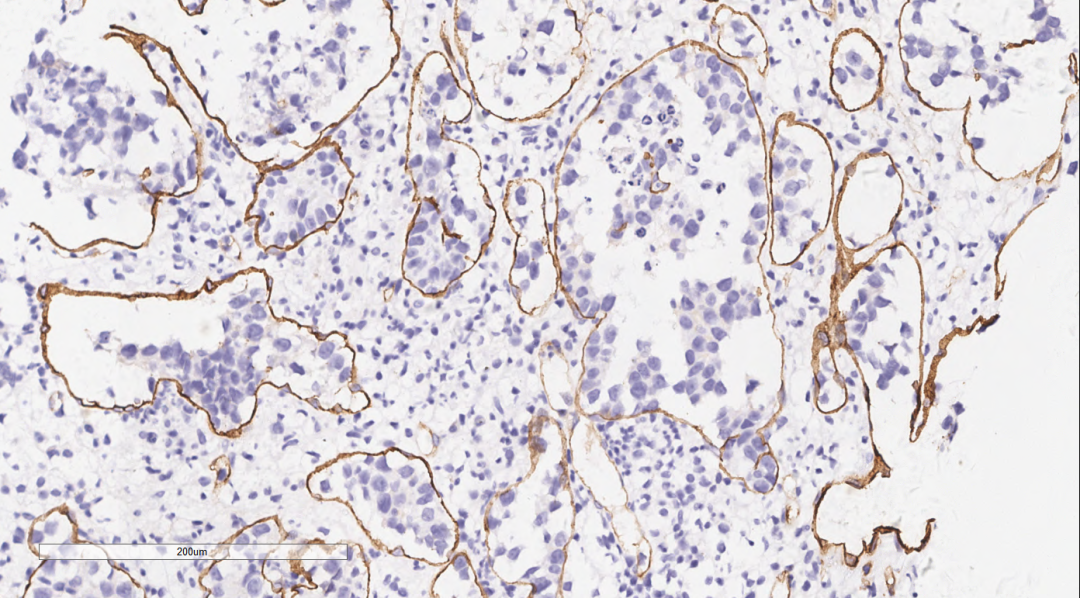
图9 肿瘤细胞完全位于CD34勾画的的血管腔隙内
石蜡切片病理诊断
肾上腺血管内大B细胞淋巴瘤。
讨论
背景:
血管内大B细胞淋巴瘤(Intravascular large B-cell lymphoma, IVLBCL)又称血管内淋巴瘤病或嗜血管性淋巴瘤,是弥漫大B细胞淋巴瘤(Diffuse large B-cell lymphoma, DLBCL)的一个罕见亚型,属于结外淋巴瘤,发病罕见。1958年Pfleger和Tappeiner首次报道该病并描述为“系统性血管内皮瘤病”。1986年,Sheibani等人确认了其是淋巴造血系统来源的恶性肿瘤,并重新定义为“嗜血管性大细胞淋巴瘤”。随后,2001年版的WHO《造血与淋巴组织肿瘤分类》中正式将该病命名为IVLBCL,并归类为结外弥漫B细胞淋巴瘤的一个独立亚型,这一分类在2022年WHO第五版的新分类中继续得到保留。
临床病理特征:
1.临床特点
IVLBCL发病率约0.5/100万,好发于中老年人,(中位年龄67~70岁,范围:34~90),男性较多见,伴有B症状(55~76%)、贫血和/或血小板减少(63~84%)、肝脾肿大(77%)、骨髓累及(75%)、血清LDH水平高(86%)和噬血细胞症(61%)。可累及不同组织和器官,以中枢神经系统、皮肤和骨髓最常见,发生在肾上腺少见。双侧肾上腺同时受累较多见,部分IVLBCL患者可伴肾上腺功能不全,发病机理很可能是肿瘤细胞易侵犯全身各个中小血管,出现的血管堵塞,导致肾上腺级全身器官缺血或梗塞。肿瘤块也可压迫肾上腺实质细胞,导致细胞萎缩和器官功能障碍。虽然肿瘤细胞位于血管内,但脑脊液及外周血常规检查常无阳性发现,在骨髓器官中受累非常轻微。病程进展迅速,以贫血和(或)血小板减少最常见。
根据临床表现,分为三种类型,分别为经典型、皮肤变异型和噬血细胞综合症相关变异型:
①经典型:
常见于西方国家,症状与主要受累器官有关。大部分患者可出现不明原因的发热、疼痛、局部器官特异性症状或多器官衰竭,或皮肤受累,并且表现出广泛的异质性病变。小部分的患者可有中枢神经系统症状,表现因受累部位不同而表现各异,类似于多灶性脑血管病变、脑梗死、脊髓和神经根病变、亚急性脑病及外周神经疾病或脑神经疾病等。
当有两种器官相关表现需考虑该病:
(1)当IVLBCL严重累及内分泌器官(主要是垂体、甲状腺和肾上腺)并导致内分泌功能不全的多种体征和症状时;
(2)当肺部受累时,影像学通常呈现毛玻璃样外观和结节。
②皮肤变异型:
主要表现为单一或多处皮肤病变,全身分期为阴性,预后好。在部分IVLBCL患者中,皮损可为首发表现,IVLBCL累及人体各部位皮肤组织的概率从高到低为下肢、躯干、上肢、臀部;皮损发生的概率从高到低为斑块/结节、斑片、毛细血管扩张、樱桃状血管瘤、紫癜。有文献显示,对于IVLBCL,即使是外观正常皮肤,也有可能发现肿瘤细胞,研究亦显示,随机皮肤盲检的阳性率高于骨髓活检。因此对于有皮损的可疑患者,推荐将皮损作为皮肤活检部位,而对于无明显皮损的可疑患者,推荐下肢和躯干部位外观正常皮肤作为活检部位。随机多部位皮肤活检(random skin biopsy, RSB)(强调必需深达肌层;建议取材部位为大腿两侧和腹部)对临床疑似患者早期明确诊断意义重大。
③噬血细胞综合症(hemophagocytic syndrome, HPS)相关变异型:
患者表现出典型的噬血细胞综合征,全身各个脏器均可侵犯,受累部位包括骨髓、脑、皮肤、肝脏、脾脏、肾脏、内分泌腺、肺、前列腺、心脏、淋巴结、胃肠道、子宫、胆囊等。主要表现为骨髓受累、发热、肝脾肿大和血小板减少症;通常伴有外周血或骨髓涂片中的非肿瘤性噬血细胞组织细胞;疾病表现出快速侵袭性发病和进展,中位生存时间为2至8个月;这种变异型主要在亚洲国家报道,尤其是日本。
2.影像学
病变早期影像学检查无特异性表现,中晚期可在影像学上表现为肿瘤占位效应,提示IVLBCL存在血管外浸润可能,并非绝对血管内生长。根据具体临床表现可以选择颅脑增强MRI、胸腹部增强 CT和正电子发射断层扫描(PET-CT)等检查。中枢神经系统受累患者的颅脑影像提示肿瘤多累及额、顶、颞、枕叶,可表现为占位效应、低密度灶或腔隙性脑梗死和出血。肺部受累患者,肺部CT可表现为双肺多发磨玻璃影、结节影和胸腔积液等。肺部易受累主要是因为:IVLBCL的病理特征为肿瘤细胞弥漫性分布于中、小血管,这种浸润病变在胸部高分辨CT检查表现为透光度下降、肺间质性病变、弥漫性磨玻璃样变而更易出现影像学变化。基于IVLBCL弥漫性浸润的特征,在影像学提示肺受累的前提下,盲检即可获得较高阳性率。在临床疑诊IVLBCL的情况下,经支气管镜肺活检(TBLB)对明确诊断帮助巨大。肾上腺受累者,CT常表现为圆形软组织密度影,病灶较小时可保持肾上腺轮廓,仅呈增生样改变,边界清晰,当肿瘤较大时可呈浸润生长,粘连或包绕邻近器官及血管,但被包绕器官的形态多无明显改变,符合淋巴瘤质地柔软、呈钻缝样生长的特性。
3.病理学特征
①镜下观:
低倍镜下瘤细胞呈单细胞或小簇状几乎全部聚集于小到中等薄壁血管腔内,毛细血管和毛细血管后微静脉多见,偶可见于大血管。肿瘤细胞存在不同的生长模式,“离散”模式:肿瘤细胞优先位于血管的中央部分并呈现自由漂浮的外观;“粘性”模式:肿瘤细胞几乎完全充满管腔,对血管结构的评估困难;“边缘化”模式:不太常见,肿瘤细胞优先粘附在内皮上,使管腔的中央部分游离。高倍镜下肿瘤细胞体积大,核/质比高,核轮廓光滑或不规则,空泡状,核仁突出,胞质稀少,核分裂象易见。偶尔也有报道出现细胞较小,染色质粗糙,或细胞核不规则病例。核仁可以是单个和突出的,或者有多个核仁,因此显示从中心母细胞到免疫母细胞/浆母细胞形态谱或间变性特征,其间变特征在HE检查中可能类似于转移癌。有些病例可能伴有充满红细胞或单核细胞的非肿瘤性组织细胞的噬血细胞现象。
②免疫组化:
IVLBCL来源于外周血B细胞,CD20、CD19、PAX5强表达,但罕见病例CD20阴性。D34和CD31标记血管内皮细胞完美勾勒出肿瘤细胞位于血管内。少数病例可能为CD30阳性。瘤细胞聚集于血管腔内的原因可能在于淋巴细胞跨血管迁移的CD29(B,-整合素)和CD54(ICAM-1)不表达、缺乏淋巴迁移所需的稳态趋化因子(Cxcr5、Ccr6、Ccr7)及异常表达G蛋白耦联受体9介导恶性淋巴细胞向血管内皮黏附,因而呈现出与其他类型淋巴瘤不同的临床表现和不良结局。文献报道大约22%~38%的IVLBCL表达CD5,尤其是噬血细胞综合症相关变异型更常见。研究显示CD5阳性的IVLBCL比CD5阴性的IVLBCL更易造成肝脾、骨髓累及,从而引发HPS,预后不佳,神经系统受累的风险较低。原发性CD5阳性DLBCI经常显示部分血管内或窦内浸润,与IVLBCL的组织学特征相似,表明CD5阳性DLBCL和IVLBCL可能有密切的相关性。有日本学者报道了皮肤CD5阳性DLBCL的病例,2年后复发为CD5阳性IVLBCL。另外,有1例发生于鼻腔的CD5阳性DLBCL的报道,化疗后完全缓解,随机的皮肤活检显示为IVIBCL,提示血管内复发,骨髓活检显示HPS,提示IVLBCL可能是CD5阳性DLBCL进展期的代表性表现。有文章显示,IVLBCL是DLBCL的一种侵袭性形式,预后极差。需要进一步研究CD5阳性 DLBCL和IVLBCL的遗传学特征,探讨CD5阳性 DLBCI和IVLBCL的发病机制,制定针对这些疾病更有效的治疗方案。有学者研究IVLBCL肿瘤细胞的分子分型,显示与非生发中心B细胞源性DLBCL的分子相似,大部分IVLBCL可能起源于生发中心后B细胞,归于非生发中心B细胞型。IVLBCL可能是DLBCL血管内复发、进展的表现。而IVLBCL是否双表达淋巴瘤(瘤细胞C-MYC≥40% 阳性和 bcl-2>50% 阳性)尚未检索出报道。Ki-67增殖指数通常在60%~100%之间,Liu等通过多因素分析发现Ki67阳性指数与预后相关。
4.分子遗传学
MIVLBCL基因组学目前研究较少,Shimada 等研究了21例IVLBCL,发现常见的遗传学改变有CD79B(67%)、MYD88(57%)、SETD1B(57%)和 HLA-B(57%)突变频率较高,存在基因相关的BCR/NFκB信号通路突变、CDKN2A/2B缺失(86%)以及PDL1/PDL2 基因拷贝数增加与异位(38%),与DLBCL的基因表达谱有一定相似性,DLBCL中存在MYD88 L265P突变,尤其在免疫豁免部位如中枢神经系统、睾丸等部位,突变率为0~94%不等,DLBCL其中MCD型遗传学表现为 MYD88和 CD79B基因突变为主,并提示MYD88基因突变在其发生、发展、治疗及预后评估中可能发挥重要作用。因此IVLBCL被认为是DLBCL的一种亚型。(但IVLBCL患者发生MYD88 L265P 和 CD79B Y196突变“双重打击”并不常见)44%~50%的病例显示PD-L1阳性,有研究结果提示PD-L1可能与IVLBCL的免疫逃逸有关。有文献报道发生在肝脏的IVLBCL伴EBER阳性的病例,并推测EB病毒感染可能与该肿瘤的预后不良相关。也有报道伴有HHV-8共感染的病例。病毒感染(如EB病毒、HVV-8)在 IVLBCL 发生中的作用,仍需进一步确认。分子遗传学检查提示IVLBCL无特异性染色体易位,瘤细胞存在免疫球蛋白轻链限制性表达和IgH基因克隆性重排。少数患者存在t(14;18)染色体异常。
鉴别诊断:
由肾上腺IVLBC穿刺活检小组织的病理诊断,因为组织小且局限,肿物压迫导致原位器官组织萎缩,极易造成漏诊与误诊,该病常需要与以下疾病鉴别:
(1)血管内NK/T细胞淋巴瘤(IVNKTL):
IVNKTL是侵袭性NK细胞白血病的一个独立的亚型,常见发生于皮肤,但也可累及其他部位。瘤细胞常为小至中等大小或小、中、大细胞混合存在,免疫组化显示肿瘤细胞表达CD3、CD56 及细胞毒分子(TIA1、GranzymeB 和 Perforin),CD5常阴性,通常不表达B细胞标志物,与EB病毒感染关系密切。IVNKTL与IVLBCL都在血管内生长,但IVLBCL不表达CD3、CD56 及细胞毒分子,表达B细胞标志物。
(2)间变性大细胞淋巴瘤(ALCL):
ALCL常见血管内弥漫大细胞浸润,但ALCL表达T细胞标志物,弥漫表达 CD30,部分ALK阳性。IVLBCL表达B细胞标志物,可表达CD30,但 ALK 阴性。
(3)反应性淋巴血管内免疫母细胞增生(ILVIP):
与IVLBCL形态学高度重叠,但存在几点不同,首先在临床特点上的不同,后者病变广泛,可累及全身各个器官,临床表现多样,ILVIP 多发生在胃肠道或阑尾,患者多表现为肠梗阻、胃肠炎症等;其次形态学的不同,ILVIP 特点是免疫母细胞样大细胞、散在小淋巴细胞及浆细胞多聚集在淋巴管/血管,大细胞可位于血管外,而IVLBCL多为形态单一大细胞特异性几乎全部位于小-中等血管内;ILVIP大细胞也可呈非GCB免疫表型,但CD30、CD38阳性,CD20、PAX5 阴性或弱阳性,IgH克隆重排阴性有助于两种肿瘤的区分。
(4)DLBCL累及血管及淋巴管:
DLBCL有时会在血管及淋巴管腔内形成瘤栓,但同时伴有明显的血管外肿块,如淋巴结受累肿大或结外脏器占位性病变,需结合临床病史、影像学检查及组织病理诊断鉴别。
(5)肾上腺皮质腺癌:
肿瘤细胞呈片状排列并被纤细的窦隙网状结构分隔,胞质透明,核异型性大,免疫组化CKpan 等上皮标记阳性。
(6)转移癌、恶性黑色素瘤及生殖细胞肿瘤:
转移癌成簇的癌细胞位于淋巴管和(或)血管内,免疫组化CK阳性而白细胞共同抗原(LCA)阴性,且临床一般有原发灶;恶性黑色素瘤瘤细胞一般弥漫成片,胞质内常可见色素颗粒,表达S-100 蛋白及 SOX10、HMB45、Melan A等,不表达B细胞标志物;生殖细胞肿瘤免疫标记SALL4、胎盘碱性磷酸酶(PLAP)、CD117和 CD30等阳性,但LCA及B细胞标志物均阴性。
治疗和预后:
目前,CHOP方案联合利妥昔单抗治疗后,患者 ORR 达到91%,3年生存率达到81%。在接受R-CHOP治疗的患者中,中枢神经系统是复发的主要部位,也是预后不良的重要因素,因此R-CHOP方案联合中枢神经系统导向治疗(例如应用甲氨蝶呤)是目前推荐的积极治疗方法。基于IVLBCL患者 MYD88、CD79B 突变频率高的特征,R-CHOP方案联合BTK抑制剂成为目前探索的方向。血管内大B细胞淋巴瘤诊治中国专家共识(2023年版)推荐对于年龄小于65岁或体能状况较好的患者行ASCT巩固治疗。一些研究发现IVLBCL患者体内程序性死亡受体配体 PD-L1、PD-L2 高表达,这为靶向治疗提供了新的靶点。近年来bcl-2抑制剂 ABT-199、XPO 抑制剂等新型药物不断出现,未来将极大改善IVLBCL患者的预后。IVLBCL 的预后主要与其累及部位相关,单纯皮肤累及患者的预后常相对较好;诊断时表现为中枢神经系统受累及合并噬血细胞综合征的IVLBCL患者预后差。IVLBCL的预后与患者治疗与否也明显相关。不良预后因素包括:年龄>60岁、未接受大剂量甲氨蝶呤治疗及未获得完全缓解。
参考文献:
[1]Ponzoni M, Ferreri A J. Intravascular lymphoma: a neoplasm of 'homeless' lymphocytes? [J]. Hematol Oncol, 2006, 24(3): 105-12.
[2]Carey R W, Harris N, Kliman B. Addison's disease secondary to lymphomatous infiltration of the adrenal glands. Recovery of adrenocortical function after chemotherapy [J]. Cancer, 1987, 59(6): 1087-90.
[3]Murase T, Yamaguchi M, Suzuki R, et al. Intravascular large B-cell lymphoma (IVLBCL): a clinicopathologic study of 96 cases with special reference to the immunophenotypic heterogeneity of CD5 [J]. Blood, 2007, 109(2): 478-85.
[4]Sheibani K, Battifora H, Winberg C D, et al. Further evidence that "malignant angioendotheliomatosis" is an angiotropic large-cell lymphoma [J]. N Engl J Med, 1986, 314(15): 943-8.
[5]陈佳梅, 罗波, 罗茜茜, et al. 我国血管内大B细胞淋巴瘤的临床亚型,诊治及预后分析 [J]. 中国癌症防治杂志, 2021, 13(3): 7.
[6]Yao X, Saad A, Chitambar C R. Intravascular large B-cell lymphoma, an exclusively small vessel disease? A case report and review of literature [J]. Leuk Res, 2010, 34(10): e275-7.
[7]中华医学会血液学分会淋巴细胞疾病学组, 中国临床肿瘤学会(CSCO)淋巴瘤专家委员会. 血管内大B细胞淋巴瘤诊治中国专家共识(2023年版) [J]. 中华血液学杂志, 2023, (03): 177-81.