
梅斯医学MedSci APP
医路相伴,成就大医



梅斯医学MedSci APP
医路相伴,成就大医
蛋白质是重要的生物大分子,在生物体内发挥着广泛的功能,了解蛋白质的三维结构对于阐明其功能和设计靶向药物至关重要。人们通常使用X射线晶体学、核磁共振(NMR)光谱学和电子显微镜等实验技术来确定蛋白质结构,但这些方法耗时长、技术难度大、成本高且适用性差。为此,科研人员开发了从头预测蛋白质结构的方法——AB Initio,利用基于物理的算法从氨基酸序列预测蛋白质结构,但其无法表征更大、更复杂的蛋白质。
近年来,机器学习算法的快速发展显著提高了蛋白质结构预测的速度,该领域最显著的成就之一是DeepMind开发的AlphaFold 2(AF2)模型,其基于深度神经网络可从氨基酸序列中预测基态蛋白质结构,耗时短并具有较高的准确性,为药物发现和基础研究开辟了新的道路、对人类健康产生了显著影响。但一系列研究发现,AF2算法在预测替代蛋白质构象和序列变异影响方面能力有限。
近期,美国布朗大学分子和细胞生物学与生物化学系的科研人员在Nature Communications上发表了题为“High-throughput prediction of protein conformational distributions with subsampled AlphaFold2”的文章。研究团队提出了一种使用AlphaFold 2通过亚采样多序列比对直接预测不同蛋白质构象相对种群的创新方法,并对两个具有截然不同可用序列数据的蛋白质进行核磁共振实验,即Abl1激酶和粒细胞-巨噬细胞集落刺激因子(GM-CSF),该方法以超80%的准确率预测了相对状态种群的变化。此外,亚采样方法在用于定性预测突变或进化对蛋白质构象分布和高密度状态的影响时效果最好。综上,该方法高效、迅速且成本较低,能有效预测蛋白质构象的相对种群,甚至可达到单点突变的分辨率,有望成为药理学、实验结果分析和预测进化的有用工具。

文章发表在Nature Communications
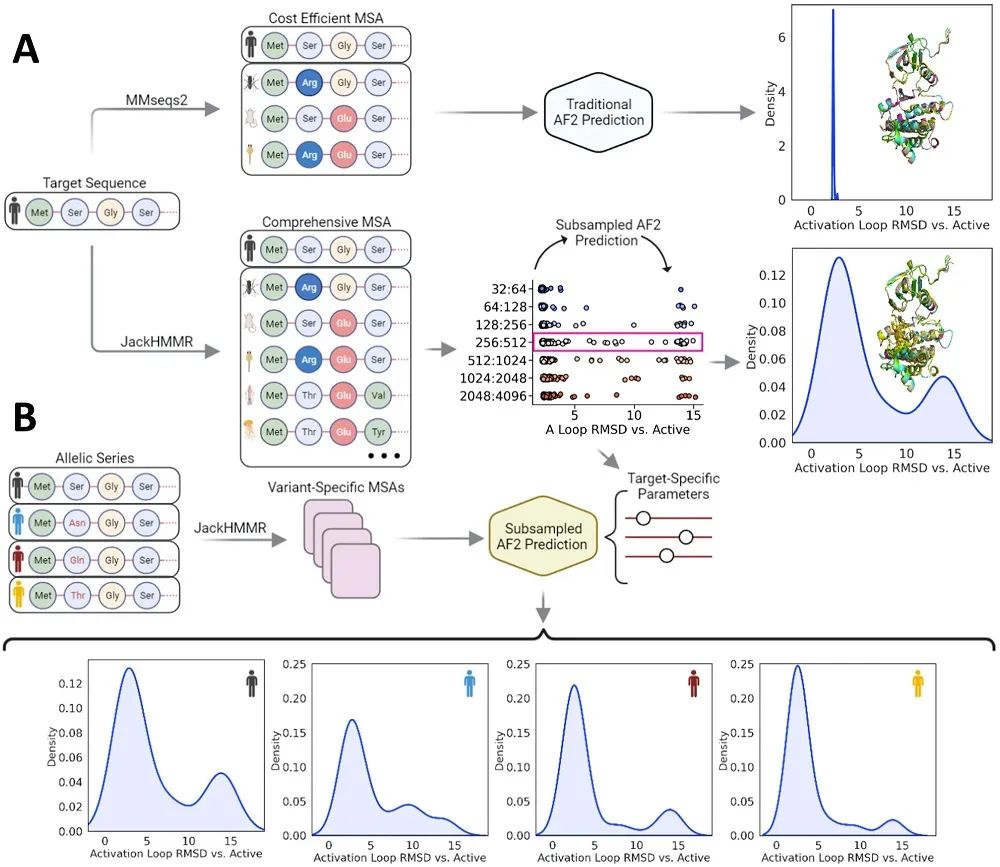
亚采样AF2工作流程
近年来,已有多个研究小组观察到,采用不同参数和多序列比对(MSA)深度的AF2方法能欧仅基于序列数据预测构象变化,这些替代AF2框架的共同原理是对MSA进行亚采样,以调节不同结构域的协同进化信号。从主MSA中随机选择任意数量的序列(由max_seq参数定义)(目标序列始终被选中),其余序列使用Hamming距离围绕每个选定序列聚类;AF2使用聚类中心和聚类中长度为extra_seq的样本进行推断(图1)。先前工作表明,在默认值的基础上显著降低max_seq值和extra_seq值,可实现对一系列模型系统的集成预测。
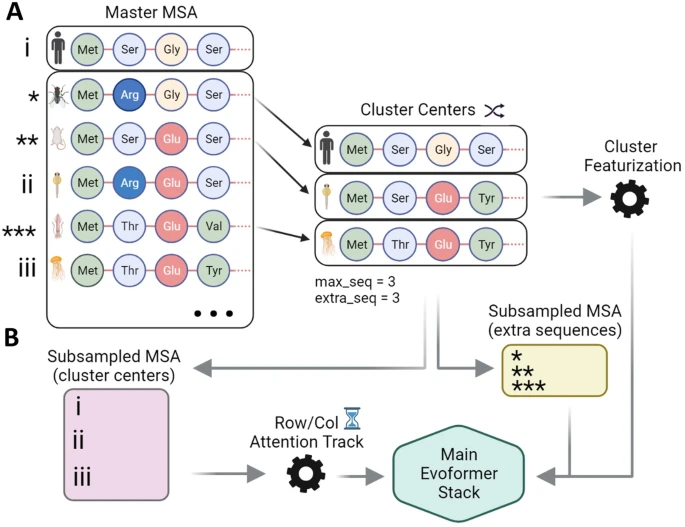
图1:AF2的MSA聚类启发式算法。
基于上述观察结果,研究团队系统测试了不同AF2参数组合预测Abl1激酶核心结构的准确性。据悉,Abl1有三种不同的主要构象:在溶液中,Abl1主要以活动(基态)状态存在,在极少数情况下会转换为非活性状态1(I1),然后转换为非活性状态2(I2);从基态到I1状态的变化较小,但从I1到I2状态的转变涉及到相当大的骨架重排。
为使用AF2生成完整的Abl1构象集合,研究团队首先通过JackHMMR算法在野生型Abl1激酶核心(残基229-515)上编译了一个涵盖60万条序列的广泛MSA;为提高结果的统计能力,为每个测试运行了32个具有独立种子的预测,并在推断过程中启用了dropout,以便从模型的不确定性中取样;所有其他参数保留默认设置。
结果显示,当max_seq:extra_seq值为256:512时,激活环构象的结果最为多样化;重要的是,AF2预测的激活环构象集合分布于Abl1从基态到I2态的转变过程中。为进一步检验AF2是否真正预测了沿转变过程的构象,研究团队将160个AF2 Abl1的亚采样预测集合与从溶液中apo Abl1的增强采样分子动力学(MD)模拟生成的I1到I2轨迹中提取的代表性快照进行比较,这一比较的代表性结果如图4所示。结果显示,AF2成功预测了Abl1激酶基态的相对种群;激活环转变的覆盖范围较大,表明使用AF2对中间状态进行采样,有可能揭示其路径和机制。
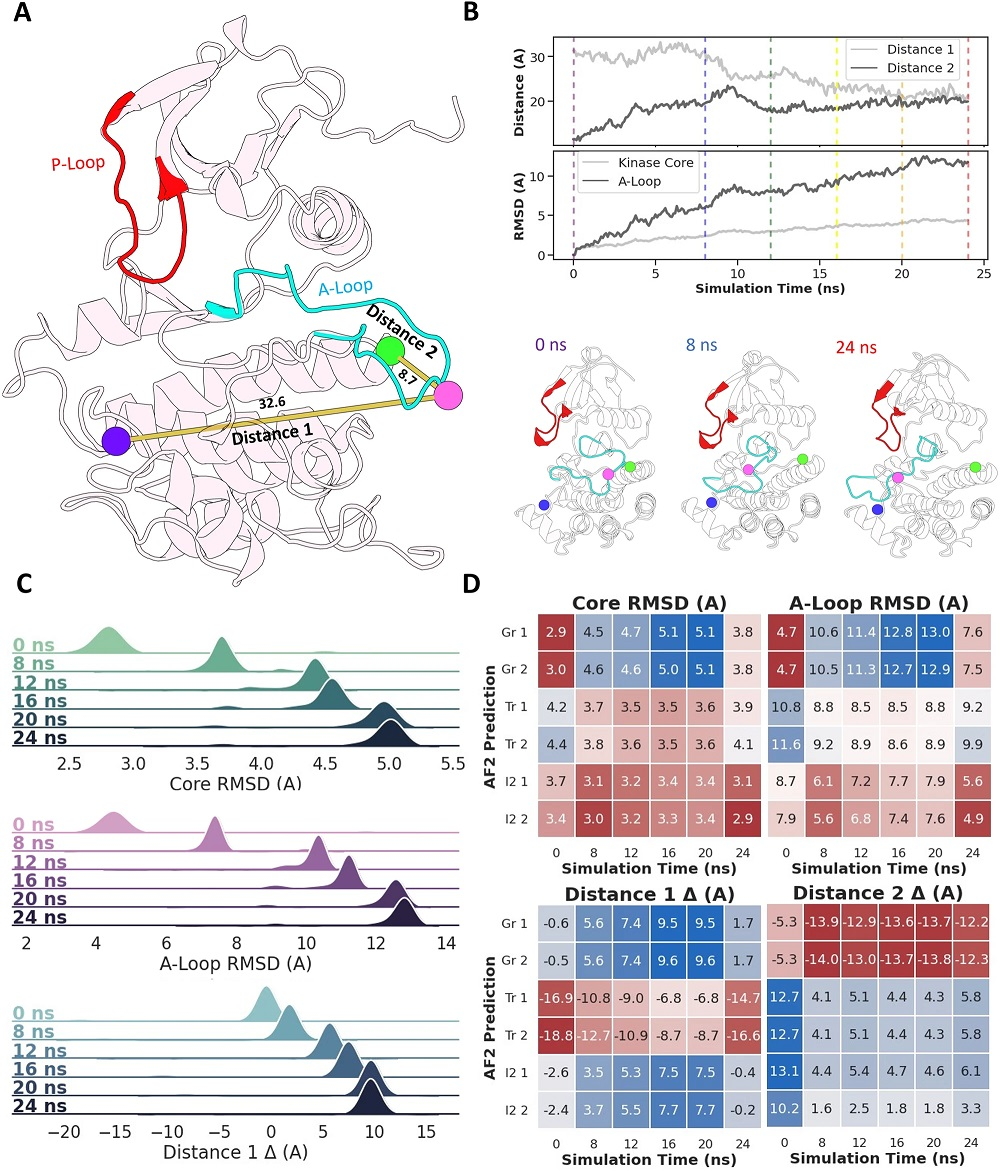
图2:通过Abl1激酶核心的增强采样MD模拟和代表性AF2预测得到的I1、I2轨迹比较。
接下来,研究团队研究了AF2在无下游MD模拟的情况下预测构象分布的潜力。已知野生型Src激酶占据基态的频率显著高于Abl1,如果亚采样AF2的假设是正确的,该方法将输出基态Src比基态 Abl1显著更多的预测。为此,研究团队使用与Abl1相同的流程为Src激酶核心(残基235-497)序列构建了一个大的MSA,并将其作为输入运行亚采样AF2,检测了Src激酶核心基态和I2态的相对种群。
结果显示,绝大多数来自亚采样AF2的Src激酶核心预测都处于基态,预测的相对状态种群为97%,而Abl1为89%。有趣的是,Src预测中没有一个被发现处于I2状态,这表明在使用AF2预测相对状态种群时存在分辨率限制:在目前的实现中,具有较低占用率的构象(如Src中的I2)可能会被算法遗漏。虽然存在分辨率问题,但亚采样AF2正确地预测了Abl1和Src激酶核心之间构象分布的差异,这证明了其作为预测相对状态种群的高通量方法的前景。研究团队还使用亚采样AF2方法对Anc-AS激酶核心(残基1-263)进行预测,并将结果与Abl1和Src病例进行比较。结果显示,预测的Anc-AS有93%的时间处于基态,介于Src(97%)和Abl1(89%)的预测频率之间,与预期结果一致。
蛋白质中的许多点突变被认为可通过改变构象和相对状态种群从而导致不同的表型(如耐药性),为此研究团队应用亚采样AF2对一系列Abl1单突变体和双突变体进行预测,这些突变体对基态和I2状态的相对种群有明显的特征和显著影响。结果显示,亚采样AF2准确预测了相对状态种群及其方向的变化,准确率超80%。
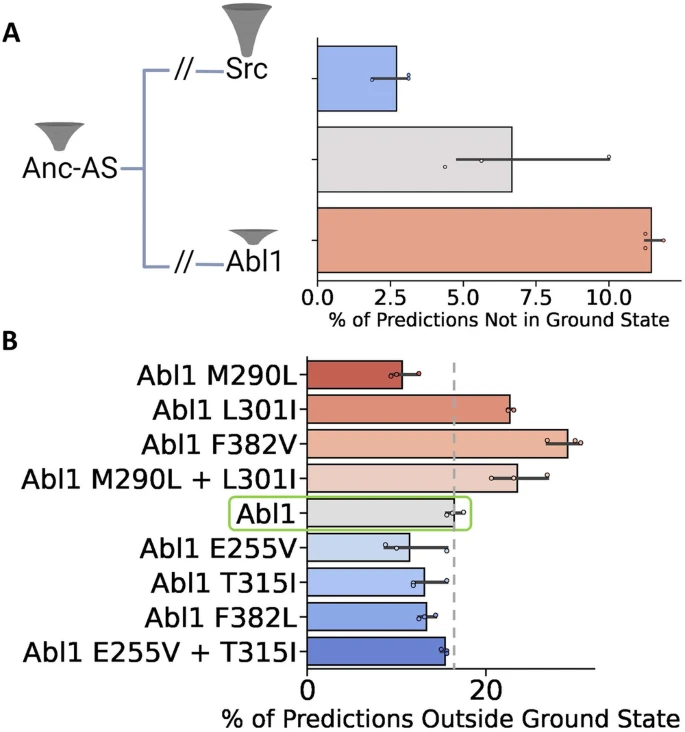
图3:亚采样AF2预测沿Src到Abl1进化途径和Abl1耐药突变的蛋白质非基态构象百分比。
考虑到对Abl1预测的成功,研究团队试图检验能否用更少的序列数据获得类似的预测结果,并使用人类GMCSF的序列重复了上述预测流程(图4)。GMCSF是一种分子量为14 kDa的单体糖蛋白,在先天性免疫中发挥核心作用,可刺激多种细胞对病原体做出应答。GMCSF中N端螺旋A的动态变化可形成两种构象:一是封闭构象(基态),这是晶体堆积时最稳定的GMCSF构象;二是开放构象,可与肝素和其他免疫系统调节剂进行结合。
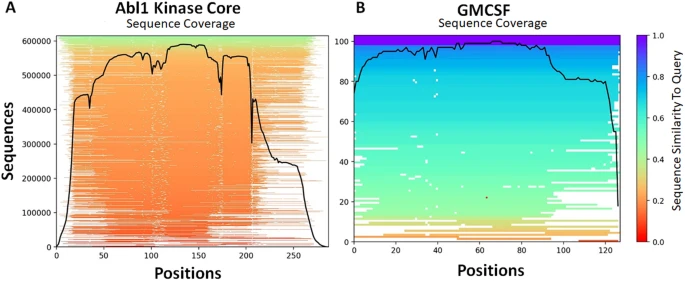
图4:该研究预测的两个蛋白质系统的构象集合。
研究团队使用野生型人类GMCSF序列作为查询和JackHMMR方法构建了MSA(112个序列)后,确定了导致GMCSF构象最大多样性的max_seq和extra_seq参数,并预测了野生型GMCSF的结构。为评估突变如何影响GMCSF的构象分布,研究团队检测了每个预测的GMCSF结构的特定主链原子位置的RMSD(图5)。
结果显示,亚采样AF2方法准确预测了突变体H15/83和H87骨架重排幅度的差异;与H87的突变相比,H15和H83的突变在构象分布方面引起的变化更大;并正确估计了突变体H83R和H83N对c端构象的显著影响,同时准确预测了H83N、H83Y和H87三种突变体对残基80-90 RMSD分布的影响。特别地,研究团队还在GMCSF中发现了一种与基态和开放态显著不同的替代构象A1,在该替代构象中,C螺旋与B螺旋交换了位置,螺旋B占据了与肝素结合的凹槽。

图5:GMCSF突变的亚采样AF2结果。
综上所述,对于Abl1激酶,亚采样AF2定性预测突变了对激酶核心活性状态种群的积极和消极影响,以及激酶核心活性到非活性转变中的大多数激活环中间状态;对于GMCSF,该方法预测了其构象集合在响应点突变时的变化,揭示亚采样AF2解码构象变化信号的卓越能力。总之,这些结果突出了AF2在预测蛋白质构象变化方面的强大且尚未开发的潜力,这将对生物物理学和药物发现领域产生重大影响。
论文原文:
Monteiro da Silva, G., Cui, J.Y., Dalgarno, D.C. et al. Author Correction: High-throughput prediction of protein conformational distributions with subsampled AlphaFold2. Nat Commun 15, 3089 (2024). https://doi.org/10.1038/s41467-024-47504-0
AlphaFold是又一个“卡”中国脖子的核心技术?“我们必须从底层技术做起”
![]() 0
2022-08-14
点击查看
0
2022-08-14
点击查看

Clinical Nutrition: 蛋白质摄入对肾移植后骨骼肌质量变化的影响
![]() 0
2022-08-19
点击查看
0
2022-08-19
点击查看
同期两篇Science:人工智能革新蛋白质设计:准确而快速地创造蛋白质
![]() 0
2022-09-24
点击查看
0
2022-09-24
点击查看
每天多25克蛋白质,可降低骨折风险!2.6万女性研究
![]() 0
2022-12-04
点击查看
0
2022-12-04
点击查看

让狼疮越来越稳定,从拒绝这种食物开始!
![]() 0
2023-01-03
点击查看
0
2023-01-03
点击查看

NEJM:食物即良药!探索蛋白质、脂肪、碳水、膳食纤维“最佳”饮食方案
![]() 0
2024-04-21
点击查看
0
2024-04-21
点击查看

