
梅斯医学MedSci APP
医路相伴,成就大医



梅斯医学MedSci APP
医路相伴,成就大医
患儿,男,10岁,“因走路不稳、头痛40余天”入院。最初家长发现患儿穿衣时动作笨拙,走路不稳。易摔倒,头痛呈阵发性,顶枕部为著,性质不详,无发热,无抽搐,无尿便障碍。既往史:出生史及生长发育史正常,学习排名中等(家长诉),患儿姐姐(16岁)、母亲、姥姥均健康,姨妈及其一子一女均健康。
入院查体:头围54 cm,血压108/70mmHg(1mmHg=0.133kPa),一般状态可,神清,爱笑,计算力差,吐字不清,周身皮肤粗糙、无色素沉着,双侧瞳孔等大等圆,对光反射灵敏,水甲眼震,咽无充血,心肺腹部查体未见异常,颈项强阴性,四肢肌力正常,肌张力高,双侧膝腱反射亢进,双侧Chaddocksign(+),昂自氏征阴性。
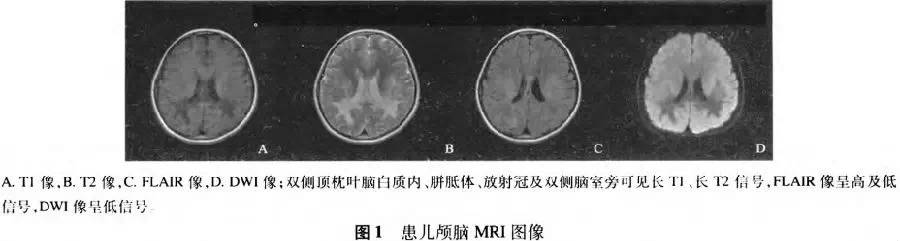
辅助检查:血尿常规、肝肾功能、心肌酶、离子、血脂、血糖、免疫三项、甲状腺功能三项、红细胞沉降率正常。血清皮质醇(08:00):185.38nmol/L(参考值240~619nmol/L),24h尿游离皮质醇301.50nmol/24h(参考值108~961 nmol/24h),血浆促肾上腺皮质激素(08:00):15.76pmol/L(参考值1.6~13.9pmol/L):脑脊液清亮,压力180 mmHg,常规生化正常,寡克隆带阴性。血清及脑脊液肺炎支原体/衣原体抗体、结核抗体阴性,单纯疱疹病毒、EB病毒、柯萨奇病毒抗体IgM阴性。胸正位片、肾上腺彩超正常。颅脑MRI(图1):双侧顶枕叶脑白质内、桥脑、胼胝体、双侧大脑脚、丘脑、基底节区、放射冠、半卵圆中心及双侧脑室旁可见长Tl、长T2信号,FLAIR像呈高及低信号,DWI像呈低信号。
智能测验:WISC-R,语言IQ65,操作IQ47,总IQ49,受试者:反应慢,注意力分散,情绪好,合作。眼底检查未见异常。ABCDI荩因测序:患儿半合子突变C.514delC,P.(Argl72Alafs*26)。母亲携带,杂合突变,患儿姐姐未携带。临床诊断为X-连锁肾上腺脑白质营养不良,建议购买Lorenzo’S油、低脂饮食和康复训练,监测皮质醇情况,必要时激素替代治疗,家K未治疗,随访1年,现患儿不能行走,吐字不清加重。
讨论
X-连锁肾上腺脑自质营养不良(X-ALD)是一种常见的过氧化物酶体脂质代谢病,呈X-连锁隐性遗传,男性受累为主,40岁以上女性携带者超过50%出现症状。在美国,男性发病率1/2l000,女性携带率1/14000,平均1/17000,我图发病率尚未明确统计。该病是由于ALD蛋白(adrenoleu-kodystrophy protein,ALDP)功能异常,引起极长链脂肪酸(very long chainfatty acids,VLCFAs)转运及β氧化障碍,进而在脑白质、肾上腺皮质等组织中大量蓄积,引起脑白质脱髓鞘和肾上腺皮质功能减退。Moser根据起病年龄、受累部位、进展速度等,将X-ALD分为以下类型:
(1)儿童脑型ALD,占35%;
(2)肾上腺脊髓神经病型ALD,占25%~46%;
(3)青少年脑型ALD,占4%~7%;
(4)成人脑型ALD,占2%~5%;
(5)单纯Addison病,占10%;
(6)无症状型,占4%~10%。其中以儿童脑型ALD和肾上腺脊髓神经病型ALD最为常见。
X-ALD具有特征性的生化与影像学改变。几乎所有男性患者及80%女性携带者可见VLCFAs增高:对于肾L腺皮质功能不全患者,24h尿皮质醇排出减少,血浆促肾上腺皮质激素升高。脑型ALD患者中,颅腑MRI异常通常早于临床症状,具有特征性改变,即腑白质呈对称性长Tl、长T2信号,约85%由后向前发展,逐一累及枕、顶、颞、额叶,可累及胼胝体压部及脑干皮质脊髓束,弓形纤维多不受累。
基因突变分析对X-ALD患者诊断有重要意义。致病基因ABCDI位于Xq28,长约21 kb,由lo个外显予和9个内含子组成,编码ALDP。目前发现的突变位点超过1 500个,其中6l%为错义突变,22%为移码突变,10%为无义突变,4%为氨基酸插入/缺失,3%为一个或多个外湿子缺失。36.9%位于外显子。同一家族后代中可出现新发突变,一些患者可同时存在多个突变位点。报道较多的突变为1415-16delAG、1202G>A、1661G>A、1553G>A,我国的热点突变位点尚不明确。
该患儿因走路不稳、动作笨拙、头痛就诊,家长诉患儿学习排名中等,但入院后发现智力落后,神经系统查体存在锥体束征,颅脑MRI改变典型,患儿无皮肤黑、失盐表现,但血清皮质醇偏低,血浆促肾上腺皮质激素偏高,临床考虑为儿童脑型ALD,未行VLCFAs检查,行ABCDI基因测序,发现突变位点,其位于外显子1b,属于移码突变,在X-Aid基因库(WWW.aid.n1)可布到该突变位点,但未见公开发表的报道,其母亲属于携带者,其姐姐未携带,患儿姨妈及子女未检测。该病缺乏特异性治疗,主要包括激素替代治疗、Lorenzo’S油、低脂饮食和造血干细胞移植,本例患儿家长放弃治疗,随访1年,病情较前加重。
